
Титульный лист и исполнители
РЕФЕРАТ
Отчет 97 с., 1 кн., 54 рис., 20 табл., 80 источников.
CRISPR, CAS9, MSTN, МИОСТАТИН, ОВЦА, ГЕНОМНОЕ РЕДАКТИРОВАНИЕ, ФИБРОБЛАСТ, ГЕН, МИКРОСКОПИЯ, СЕКВЕНИРОВАНИЕ
В отчете представлена информация по структуре генома овец, полученная при изучении источников иностранной литературы и баз данных, информация по методам оценки мясной продуктивности овец и существующим модификациям технологии редактирования генома методом CRISPR/Cas с учетом возможности их применения у овец. В ходе работы подготовлена материально-техническая база для проведения экспериментов по редактированию генома, сконструированы гайдовые последовательности и плазмидные векторы для направленного редактирования первого экзона гена миостатина овец. Проведено клонирование генных конструкций в компетентных клетках и секвенирование их последовательностей. Получены культуры фибробластов от овец северокавказской мясошерстной породы. С использованием физических и химических методов разработанные генные конструкции внесены в фибробласты овцы. Получены культуры клеток с отредактированным геномом. Модифицированные клетки были оценены с использованием световой и люминесцентной микроскопии, а также по результатам секвенирования выделенных ДНК. С учетом результатов секвенирования используемых векторов, анализа показателей жизнеспособности модифицированных фибробластов и их люминесцентной микроскопии, результатов секвенирования ДНК, выделенных из редактируемых клеток установлено, что обе разработанные CRISPR-кассеты являются работоспособными и пригодны для редактирования клеток овец. Однако, наибольшей эффективностью обладает конструкция 222.
ВВЕДЕНИЕ
Система CRISPR/Cas9 – современный, мощный и точный инструмент для геномного редактирования. Впервые возможность использования системы адаптивного иммунитета бактерий CRISPR/Cas9 для направленной модификации генома клеток млекопитающих была продемонстрирована в 2013 году [1–3]. Относительная простота подбора, синтеза и использования системы CRISPR/Cas9, высокая точность и эффективность привели к ее широкому использованию в самых различных направлениях молекулярной биологии в том числе для нокаута генов влияющих на продуктивность сельскохозяйственных животных [4]. Одним из лидеров в области CRISPR/Cas редактирования является Китай. Именно в Китае в 2014 году с применением системы CRISPR/Cas9 были получены первые клинически здоровые ягнята с отредактированной последовательностью гена миостатина (GDF-8, MSTN, TGF-8) – одного из ключевых регуляторов мышечного роста, ограничивающего дифференцировку и пролиферацию миосателлитов, миобластов и некоторых других видов клеток [5–8]. К 2018 году с использованием системы CRISPR/Cas9 учеными из Китая были получены первые ягнята нуль-мутанты по гену миостатина обладающие фенотипом двойной мускулатуры [9,10]. Аналогичные эксперименты проводятся на козах, свиньях и животных других видов по всему миру [11–14].
Актуальность вопросов получения CRISPR/Cas модифицированных клеточных линий, эмбрионов и животных, обусловлена не только общемировым научным интересом, но и перспективами практического внедрения технологии в сельское хозяйство. С использованием системы CRISPR/Cas9 возможно получить животных с уникальными показателями продуктивности как по количественным, так и по качественным показателям. В связи с этим целью наших исследований явилась разработка методов использования технологии редактирования генома CRISPR/CAS для повышения продуктивности сельскохозяйственных животных на примере овец российских пород.
1 Изучение источников иностранной литературы и баз данных (NCBI, ENSEMBL и т.п.) с информацией о структуре генома овец
Первая сборка генома овцы – Ovis_aries_1.0 была представлена Международным консорциумом по геномике овец (International Sheep Genomics Consortium, ISGC) в международной базе данных Национального центра биотехнологической информации (National Center for Biotechnology Information, NCBI) в 2010 году. Сборка генома была сформирована с трехкратным покрытием по результатам секвенирования геномов шести самок овец с использованием секвенатора Roche FLX 454. В исследовании были использованы мериносовые и помесные овцы, овцы породы полл дорсет (Poll Dorset) и авасси (Awassi). Контиги были собраны на основе выравнивания по бычьему геному. Сборка содержала данные по 20 921 белок-кодирующим генам и 290 псевсдогенам.
В 2012 году ISGC представил сборку Oar_v3.1, созданную по результатам секвенирования геномов овцы и барана породы тексель [15]. Секвенирование проводили с использованием технологии Illumina GAII. Для «заполнения пробелов» использовали данные сборки Ovis_aries_1.0. В 2015 году эта сборка была обновлена до версии Oar_v4.0. Сборка содержит данные по 20 645 белок-кодирующим генам и 3 224 псевдогенам [16].
В 2017 году была начата работу по подготовке новой сборки генома овцы. В 2019 году был опубликован репрезентативный геном Oar_rambouillet_v1.0, созданный специалистами Центра секвенирования генома человека Бэйлорского колледжа медицины (Baylor College of Medicine Human Genome Sequencing Center). Секвенирование выполнялось с использованием технологий HiSeq X Ten и PacBio RS II, для анализа было использованы образцы от 52 овец породы рамбулье (Rambouillet). Сборка содержит данные по 21 160 белок-кодирующим генам и 5 223 псевдогенам. На данный момент версия Oar_rambouillet_v1.0 является наиболее полной и актуальной.
Домашняя овца (Ovis aries) имеет 26 пар аутосом и две половые хромосомы: X и Y. Наибольшее количество белок-кодирующих последовательностей согласно обнаружено на хромосомах 1, 2 и 3, меньше всего на 24 и 26 (Таблица 1).
Распределение функционально значимых последовательностей, кодирующих белки и другие молекулы по этим хромосомам схематично предоставлено на рисунках 1,2,3, 4 и 5.
Таблица 1 – Распределение кодирующих элементов в геноме овцы
| Хромосома | Размер (Mb) | Белки | Рибосомальные РНК | Транспортные РНК | Другие РНК | Гены | Псевдогены |
|---|---|---|---|---|---|---|---|
| 1 | 301.31 | 4223 | — | 294 | 738 | 3430 | 541 |
| 2 | 265.69 | 3168 | 2 | 108 | 536 | 2386 | 380 |
| 3 | 241.14 | 4268 | — | 123 | 675 | 3077 | 454 |
| 4 | 130.07 | 1286 | — | 53 | 203 | 1112 | 194 |
| 5 | 117.63 | 2461 | 2 | 84 | 333 | 1844 | 274 |
| 6 | 129.79 | 1245 | — | 47 | 232 | 950 | 177 |
| 7 | 107.7 | 1616 | — | 52 | 284 | 1412 | 226 |
| 8 | 98.77 | 850 | — | 47 | 181 | 783 | 176 |
| 9 | 104.71 | 1076 | — | 47 | 198 | 783 | 112 |
| 10 | 97.21 | 665 | 1 | 33 | 162 | 611 | 114 |
| 11 | 60.98 | 2420 | — | 81 | 384 | 1641 | 130 |
| 12 | 84.52 | 1189 | — | 48 | 244 | 920 | 109 |
| 13 | 87.26 | 1558 | — | 37 | 288 | 1075 | 131 |
| 14 | 71.11 | 2267 | — | 56 | 372 | 1730 | 210 |
| 15 | 90.32 | 1723 | — | 44 | 154 | 1482 | 347 |
| 16 | 78.35 | 544 | — | 20 | 132 | 504 | 102 |
| 17 | 82.58 | 1196 | — | 48 | 198 | 889 | 121 |
| 18 | 70.86 | 916 | 1 | 31 | 473 | 940 | 133 |
| 19 | 62.71 | 1247 | — | 25 | 246 | 750 | 75 |
| 20 | 55.94 | 1371 | — | 203 | 248 | 1253 | 141 |
| 21 | 52.95 | 1264 | — | 22 | 175 | 942 | 160 |
| 22 | 55.67 | 851 | — | 22 | 122 | 544 | 73 |
| 23 | 68.31 | 641 | — | 21 | 147 | 458 | 71 |
| 24 | 43.08 | 1357 | — | 64 | 182 | 927 | 75 |
| 25 | 47.66 | 635 | 13 | 20 | 132 | 457 | 66 |
| 26 | 49.39 | 437 | — | 20 | 109 | 368 | 56 |
| X | 153.34 | 1716 | — | 85 | 307 | 1551 | 356 |
| MT | 0.02 | 13 | 2 | 22 | — | 13 | — |
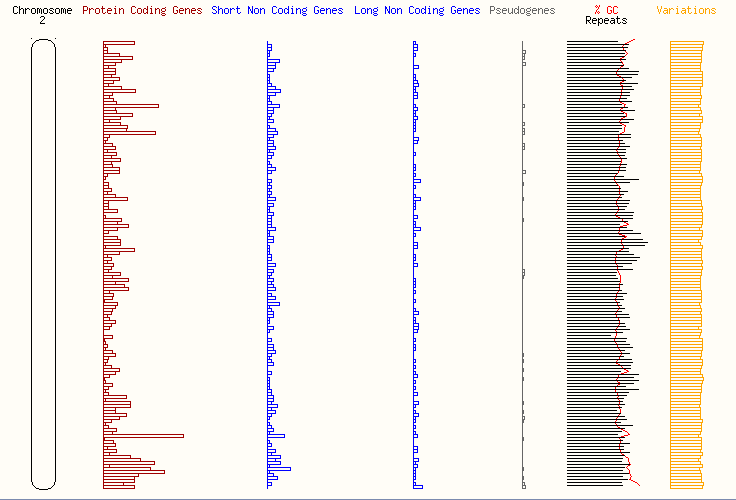
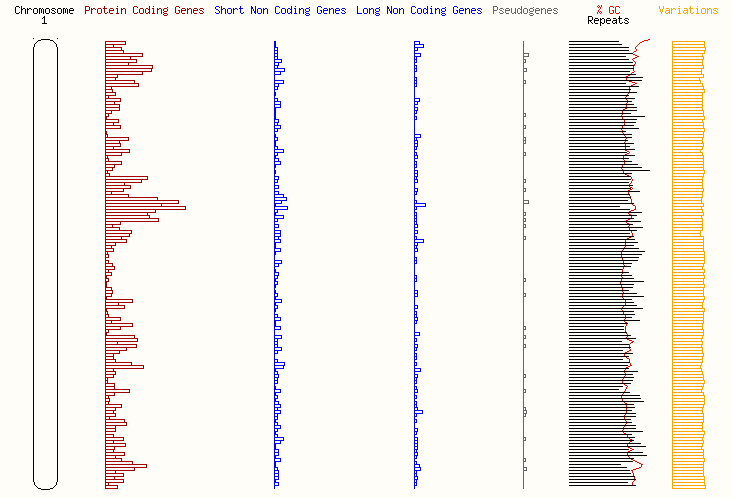 Рисунок 1 – Распределение генов на хромосоме 1
Рисунок 1 – Распределение генов на хромосоме 1
Рисунок 2 – Распределение генов на хромосоме 2
Рисунок 3 – Распределение генов на хромосоме 3 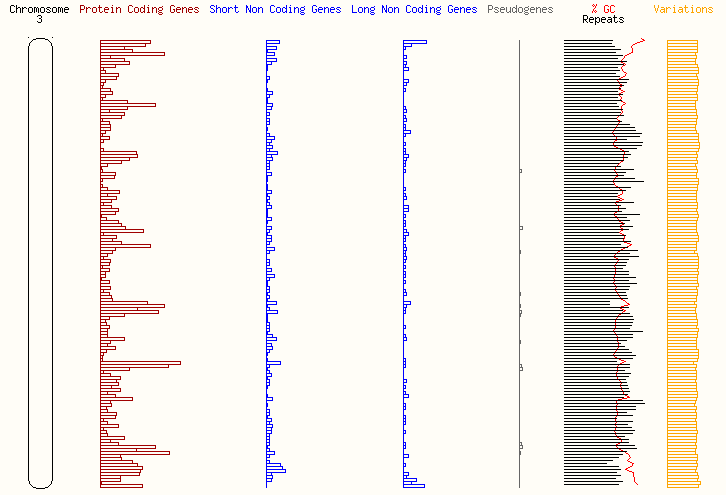
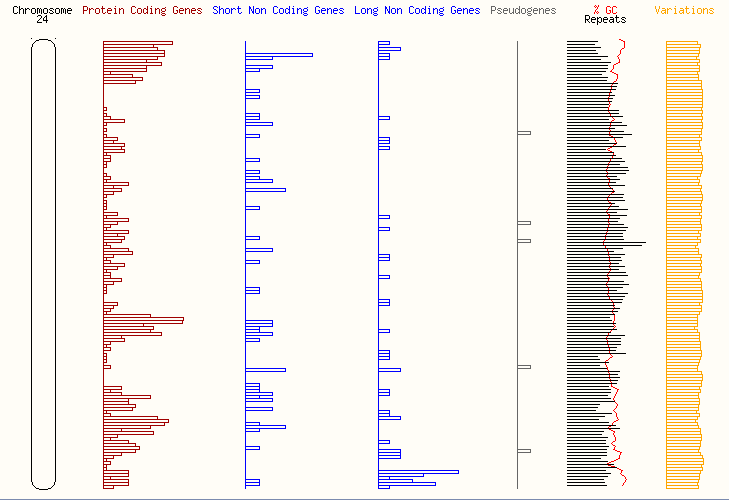
Рисунок 1 – Распределение генов на хромосоме 24
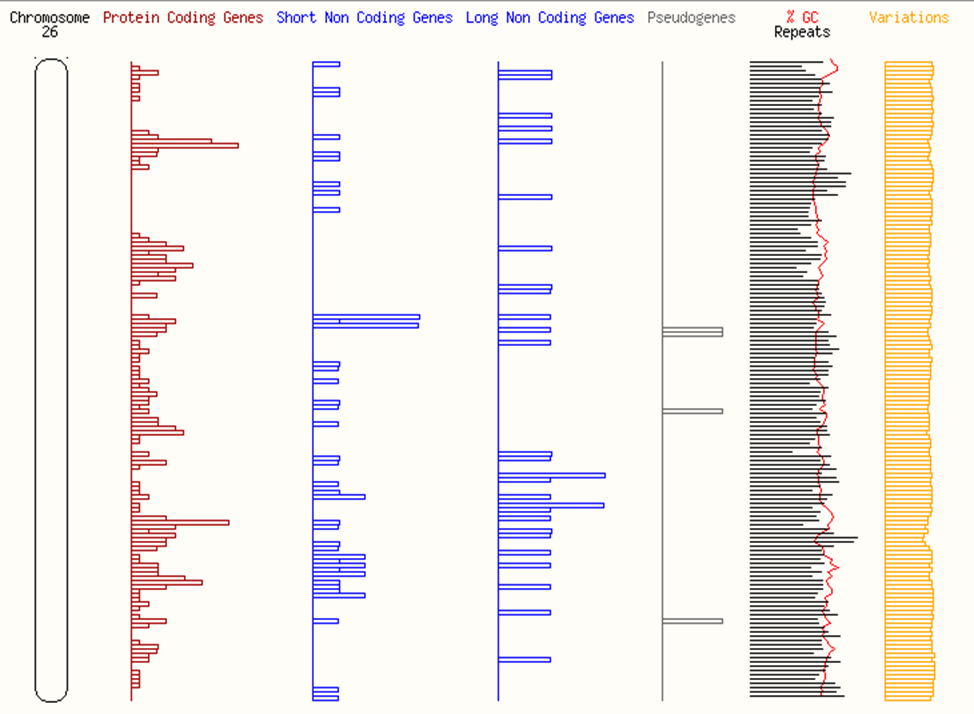
Рисунок 2 – Распределение генов на хромосоме 26
В базах данных также представлена информация по последовательностям митохондриальной ДНК. В митохондриальной ДНК обнаружено 13 белок-кодирующих генов (Рисунок 6) [17,18] .
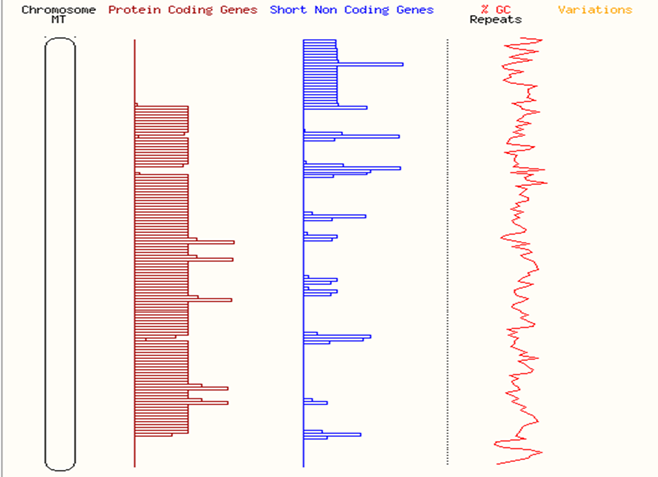
Рисунок 3 – Распределение генов в митохондриальной ДНК
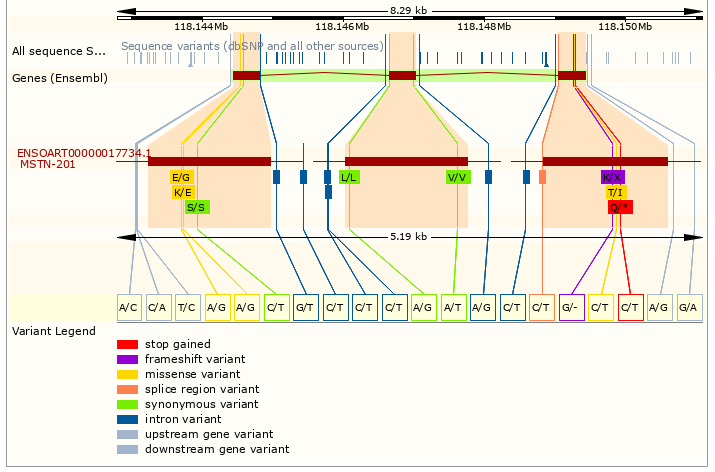 В геноме овцы обнаружено более 61 000 000 различных полиморфизмов. В том числе в области генов, регулирующих рост и развитие организма. Так в области гена миостатина (MSTN), ограничивающего пролиферацию и дифференцировку мышечных клеток выявлено 188 полиморфизмов [19–23]. Большинство из них расположено в некодирующих областях гена и не влияет на конечную аминокислотную последовательность белка [24,25]. В области экзонов выявлено 8 однонуклеотидных замен (Рисунок 7) [26–28].
В геноме овцы обнаружено более 61 000 000 различных полиморфизмов. В том числе в области генов, регулирующих рост и развитие организма. Так в области гена миостатина (MSTN), ограничивающего пролиферацию и дифференцировку мышечных клеток выявлено 188 полиморфизмов [19–23]. Большинство из них расположено в некодирующих областях гена и не влияет на конечную аминокислотную последовательность белка [24,25]. В области экзонов выявлено 8 однонуклеотидных замен (Рисунок 7) [26–28].
Рисунок 7 – Однонуклеотидные замены в области экзонов
Из рисунка 7 видно, что несинонимичные мутации расположены в области экзонов 1 и 3. Три из представленных замен приводят к изменению аминокислоты, одна к сдвигу рамки считывания и одна к преждевременному образованию стоп-кодона.
Ген MSTN у овец расположен на второй хромосоме, имеет 3 экзона и 2 интрона. Зрелая мРНК гена MSTN состоит из 1128 нуклеотидов. Ген кодирует пептид, состоящий из 395 аминокислот.
Уникальной особенностью овец является наличие у них альтернативного варианта сплайсинга пре-мРНК миостатина (myostatin splice variant — MSV) [29].
Полипептидная последовательность, кодируемая мРНК MSV содержит N-концевой домен с 256 аминокислотами, идентичный каноническому миостатину, и уникальный C-конец из 65 аминокислот. Ученые обнаружили, что в гене MSTN овец имеется скрытый интрон, охватывающий 1011 нуклеотидов, расположенных в области третьего экзона и 3’ фланкирующей области гена. Альтернативный сплайсинг путем добавления новой C-концевой кодирующей последовательности к усеченной кодирующей последовательности пропептида миостатина, состоящей из экзонов 1, 2 и части экзона 3, создает в транскрипте MSV новую открытую рамку считывания длиной 966 пар оснований. Матричная РНК MSV трансформируется в белок, который в скелетных мышцах функционирует как антагонист канонического миостатина. Белок MSV стимулирует миогенез посредством индукции миогенных регуляторных факторов. Избыточная экспрессия MSV сопровождается увеличением количества синтезируемых белков MyoD, Myogenin и MRF4. MSV представляет собой уникальный пример внутригенной регуляции, при которой альтернативный вариант кодируемого белка непосредственно снижает биологическую активность канонического продукта гена [29,30].
У овец ген MSTN и прилегающие области входят в локусы количественных признаков (QTL) связанные с интенсивностью мышечного роста и выходом баранины [31–36], с содержанием в мясе ряда жирных кислот, таких как арахидоновая, эйкозапентаеновая, линоленовая, докозапентаеновая [37]. Помимо этого, в области гена MSTN обнаружены QTL, ассоциированные с молочной продуктивностью [38] и с устойчивостью к паразитарным заболеваниям [39,40].
В последовательности гена миогенной дифференцировки-1 (MyoD1), контролирующего работу гена MSTN выявлено 85 полиморфизмов, в том числе 19 SNP в кодирующей области (Рисунок 8) [41,42].
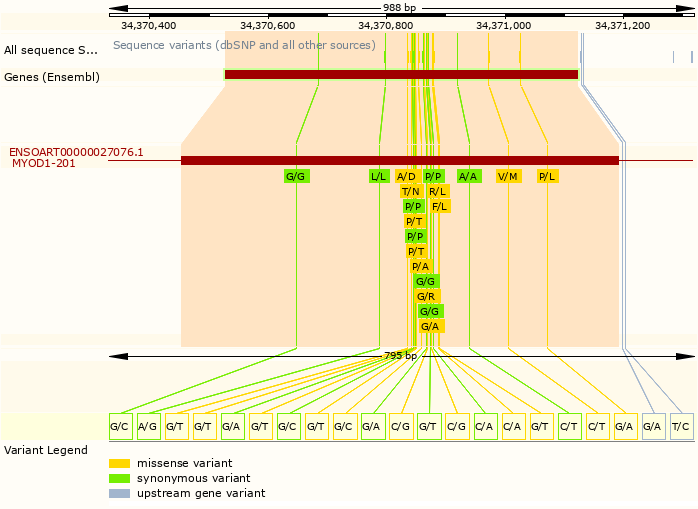
Рисунок 8 – Однонуклеотидные замены в области гена MyoD1
Одиннадцать однонуклеотидных замен приводят к изменению аминокислотной последовательности.
Общая протяженность гена MyoD1 у овец составляет 2763 нуклеотида. В состав первого экзона входит 5’UTR не транслируемая область, протяженность которой составляет 342 нуклеотида. Несмотря на то, что 5’UTR область является не транслируемой, она содержит старт транскрипции. Это является особенностью гена MyoD1, а также некоторых других генов. Протяженность кодирующей части первого экзона составляет 592 нуклеотида. Во втором экзоне содержится 78 нуклеотидов. В третьем экзоне содержится 980 нуклеотидов, из которых 264 нуклеотида расположены в кодирующей части, остальные 716 нуклеотидов расположены в не кодирующей 3′ UTR области. В первом интроне расположено 505 нуклеотидов, а во втором – 266 нуклеотидов.
В общей сложности в геноме овцы обнаружено 3 000 локусов количественных признаков (Quantitative trait locus, QTL), связанных с 256 различными параметрами (https://www.animalgenome.org). Наибольшее количество выявленных локусов количественных признаков связано с показателями молочной продуктивности, так с жирностью молока ассоциировано 450 локусов, с количеством получаемого молока – 233 локуса, с содержанием белка в молоке – 159. С параметрами мясной продуктивности связано 348 QTL, влияющих на рост овец, 229 QTL влияющих на формирование их анатомических особенностей. Помимо этого, на качество мяса влияет 20 QTL ассоциированных с текстурой мяса, 6 QTL связанных с его цветом и 61 QTL связанный с содержанием в мясе жирных кислот.
На рисунке 9 показано как распределены по хромосомам овец 63 локуса количественных признаков, связанных с живой массой.
Наличие большого количества QTL говорит о значительных перспективах для разработок в области геномного редактирования и маркер-ассоциированной селекции [43,44]. Из 61 миллиона полиморфизмов, разбросанных по всему геному. необходимо выделить каузальные мутации, связанные с формирование 3000 локусов количественных признаков и определить механизм их влияния на продуктивные показатели. Располагая достаточной информацией о функциональной роли и строении определённых участков генома, можно провести их натравленное редактирование для получения животных с интересующими характеристиками.
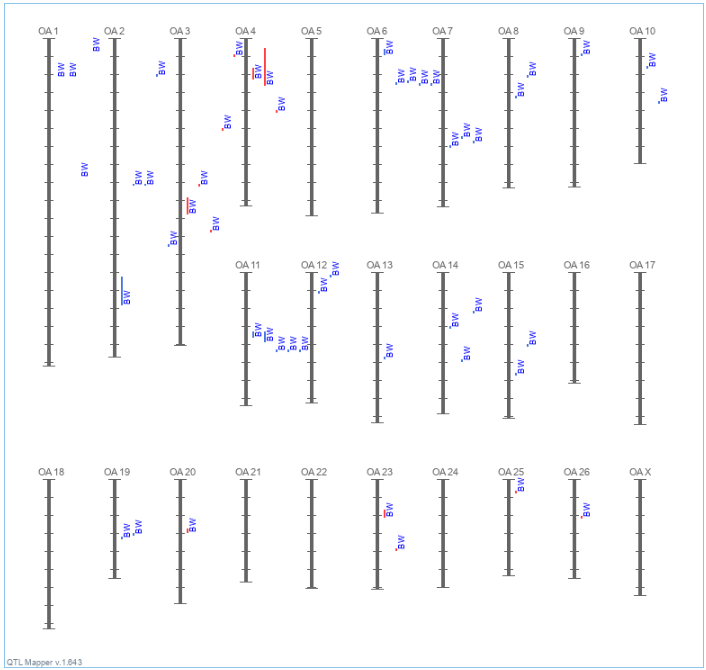
Рисунок 9 – Локализация QTL ассоциированных с живой массой
2. Изучение методов оценки мясной продуктивности овец
На современном этапе развития животноводства одна из самых важных и сложных проблем — увеличение производства мяса. Мясная продуктивность обусловлена морфологическими и физиологическими особенностями животных, формирующимися под влиянием наследственности, условий кормления и содержания. характеризуется количественными и качественными показателями. Количественными показателями являются живая и убойная масса, убойный выход, абсолютный, относительный и среднесуточный прирост. К качественным показателям относят морфологический состав туши, химический состав, калорийность, биологическую полноценность и вкусовые свойства мяса. Количественные показатели мясной продуктивности зависят главным образом от условий кормления и содержания. На качественные показатели, помимо этих условий, в значительной степени влияют породные особенности животных, пол и возраст. Очень важным показателем оценки мясной продуктивности скота является морфологический состав туши.
Мясную продуктивность овец оценивают при жизни и после убоя животных путем взвешивания, внешнего осмотра и определения упитанности. При жизни мясную продуктивность овец оценивают по целому ряду показателей.
Живая масса определяется путем взвешивания. Взвешивают животных индивидуально до кормления. Ягнят взвешивают при рождении, при отъеме от маток (4–5 месяцев), перед реализацией на убой (возраст до 1 года) или на племя. Передержка баранчиков (кроме племенных) часто бывает неоправданной как с экономической, так и биологоэкологической стороны. Необходимо иметь в виду, что обычно самая низкая себестоимость мяса получается при реализации молодняка с хорошей кондицией именно в первый год жизни. Биологический фактор проявляется тем, что у улучшенных по мясной продуктивности пород овец все стадии развития их организма проходят быстрее. Наибольшее количество самой ценной мышечной ткани у них накапливается в первый год жизни, и в этот период практически заканчивается формирование мясной продуктивности. Живая масса взрослых маток определяется осенью перед случкой, производителей — весной при бонитировке и осенью перед случкой. Ярок и баранчиков тонкорунных, полутонкорунных и жирнохвостых пород взвешивают при бонитировке в возрасте 12 месяцев, а курдючных — в 18 месяцев. На основании результатов взвешивания рассчитывают абсолютный, среднесуточный, относительный прирост.
Предубойная живая масса — масса животного перед убоем после 24‑часовой голодной выдержки.
Категории упитанности овец устанавливаются на основе степени развития мышечной и жировой ткани на холке, спине, пояснице, ребрах и у корня хвоста, а у курдючных и жирнохвостых овец — курдюка или жирного хвоста.
После убоя мясную продуктивность овец оценивают по следующим показателям.
Масса туши — туша убитого животного с почками и околопочечным жиром, но без внутренних органов, головы, хвоста, ног, кожи — определяется путем взвешивания на весах с точностью до 0,01 кг. При этом от туловища отделяют передние ноги по запястному суставу, задние — по скакательному. Сразу после убоя определяют массу парной туши, а спустя 24 часа после ее остывания в холодильной камере при температуре 4–6°С — массу охлажденной туши. На массу туши оказывает влияние порода, пол, возраст, упитанность животного.
Убойная масса включает массу туши и внутреннего жира (сальникового, желудочного, кишечного) и определяется путем взвешивания составных частей. В убойную массу у овец мясосальных и жирнохвостых пород включают массу курдюка и жирного хвоста, которые при убое отделяются от туши и учитываются отдельно. Масса туши и масса жира учитываются раздельно.
Убойный выход — это отношение убойной массы к предубойной живой массе животного, выраженное в процентах. Этот показатель колеблется в пределах от 35% до 60% в зависимости от породы, упитанности, пола, возраста животных и т. д.
Морфологический состав туши — это процентное отношение массы отдельных тканей (мышечной (35–40 %), жировой (6–24 %), соединительной (6– 8 %) и костной (14–22 %) к общей массе туши. Соотношение основных частей туши обуславливает ее пищевую ценность и зависит от породы, возраста, пола и упитанности животных.
Коэффициент мясности — это отношение в туше массы мякоти к массе костей. У овец он составляет 1:3–1:5. Площадь поперечного сечения длиннейшей мышцы спины («мышечный глазок») тесно связана с мясностью туши. Положительная корреляция между массой мышц в туше и площадью мышечного глазка у мясошерстных ягнят составила 0,77–0,81. Поэтому о мясности туши можно судить и по площади поперечного сечения длиннейшей мышцы спины. У скороспелых мясных пород овец площадь мышечного глазка больше, чем у мериносов.
Химический состав мяса и калорийность характеризуется содержанием воды, белка, жира, минеральных веществ
3. Изучение существующих модификаций технологии редактирования генома методом CRISPR/CAS, сравнение их возможностей применения у овец
Впервые возможность использования системы адаптивного иммунитета бактерий CRISPR/Cas9 для направленной модификации генома клеток млекопитающих была продемонстрирована в 2013 году. У бактерий и архей система CRISPR/Cas обеспечивает уничтожение чужеродной ДНК при инфекции фаговыми частицами. Деградацию чужеродной ДНК обеспечивает комплекс некодирующих РНК и белков, в котором за узнавание мишени отвечает «направляющая» РНК, по принципу комплементарности связывающая целевую ДНК. Кодируется данная система локусом CRISPR, экспрессирующим некодирующую РНК, и генами cas (CRISPR-associated), продуктом которых являются белки Cas. Локус CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) представляет собой кластер коротких палиндромных повторов (30-40 п.н.), разделенных участками уникальной ДНК – спейсерами (20-80 п.н.). Спейсеры локуса CRISPR гомологичны последовательностям ДНК различных фагов и различных плазмид [45] и обеспечивают специфичность действия системы [46]. К локусу CRISPR примыкает лидерная последовательность (длиной до 550 п. н.), играющая роль промотора, обеспечивающего однонаправленную транскрипцию повторов и спейсеров. Интенсивность транскрипции увеличивается в стрессовых условиях, например, при столкновении клетки с фагами [47] .
Существует большое разнообразие генов Cas [48]. Современная классификация включает пять типов систем CRISPR/Cas (I-V), объединенных в два класса, и больше десяти подтипов и основана на выявленных характерных элементах организации оперонов Cas.
В геномной инженерии, наиболее востребованной стала CRISPR/Cas-система типа II-А, выделенная из бактерии Streptococcus pyogenes. Преимущество этой системы для редактирования геномов эукариот заключается в том, что внесение двуцепочечных разрывов (ДЦР) в целевой сайт обеспечивается комплексом некодирующих РНК и всего одного крупного полифункционального белка – Cas9. В системах II-A типа белок Cas9 осуществляет процессинг pre-crRNA и интерференцию чужеродной ДНК [49].
У бактерий S. pyogenes в результате транскрипции локуса CRISPR образуется длинная некодирующая пре-crРНК, состоящая из повторов и уникальных последовательностей. С последовательностями повторов в pre‑crRNA, формируя дуплекс, комплементарно связываются молекулы малой некодирующей РНК – tracrRNA (trans-activating crRNA; трансактивирующая crРНК). Молекула tracrРНК имеет на 3’-конце три шпильки, две из них обеспечивают связывание с белком Cas9, стабилизирующим комплекс tracrРНК-пре-crРНК [50,51]. После формирования дуплеса, двухцепочечную РНК комплекса tracrРНК- пре-crРНК разрезает рибонуклеаза клетки-хозяина – РНКаза III. При этом образуются более короткие фрагменты, которые затем снова подвергаются процессингу в комплексе с tracrРНК и Cas9 [3].
Образованная зрелая crРНК содержит на 5′-конце 20-нуклеотидную спейсерную последовательность, на 3’- конце – 19-22 нуклеотидов повтора и направляет комплекс tracrРНК-crРНК-Cas9 к протоспейсеру в чужеродной ДНК. Образование комплекса Cas9-tracrРНК-crРНК, приводит к активации белка Cas9 и в случае обнаружения комплексом целевого протоспейсера с мотивом PAM (мотив, прилегающий к протоспейсеру, protospacer-adjacent motif – NGG) на 3’-конце запускается процесс интерференции – деградации чужеродной ДНК.
Активированный белок Cas9 «сканирует» ДНК, узнает PAM. Далее за счет геликазной активности Cas9 происходит расплетание участка ДНК с 5’‑конца от PAM. В случае комплементарности с crРНК происходит образование гетеродуплекса протоспейсер-crРНК. При успешной гибридизации первых восьми нуклеотидов с 3’-конца протоспейсера с crРНК происходит дальнейшая гибридизация в 5’-направлении. При успешном связывании протоспейсер-crРНК нуклеазные домены белка Cas9 вносят разрывы: в вытесненную некомплементарную цепь и цепь, с которой комплементарно связывается crРНК [52,53]. Чужеродный генетический материал разрушается.
В 2013 году впервые была продемонстрирована возможность использования системы адаптивного иммунитета бактерий CRISPR/Cas9 для направленной модификации генома клеток млекопитающих [1]. Был разработан ряд плазмидных векторов, обеспечивающих экспрессию элементов, необходимых для функционирования системы CRISPR/Cas. При помощи стандартных методов генной инженерии в такой вектор клонируют спейсерную последовательность, программирующую эффекторный комплекс на разрезание целевой протоспейсерной последовательности и редакцию интересующих участков ДНК. При редактировании генома млекопитающих результатом работы CRISPR-Cas9 является не разрушение всей молекулы ДНК, а репарация двуцепочечного разрыва, произведённого Cas9. Репарация может проводиться как за счёт негомологичного соединения концов, так и путём гомологичной рекомбинации. В результате репарации, сопровождавшейся негомологичным соединением концов, часто возникают небольшие вставки или делеции, способные разрушить рамку считывания белок-кодирующих генов, что приводит к утрате функции гена-мишени. Вызвав множество двухцепочечных разрывов, можно добиться появления крупных делеций и даже инверсий [49,54,55].
В генной инженерии используется упрощенный вариант и без того компактной системы CRISPR/Cas9. Эта система функционирует в виде комплекса из белка Cas9 и единой химерной направляющей/гидовой РНК – sgРНК. Используемая в такой системе нуклеаза Spcas9 (Cas9 S. pyogenes) адаптирована для адекватной транскрипции в клетках высших эукариот и оптимизирована по кодонам. Для обеспечения ядерной компартментализации в вектор дополнительно включены сигнальные последовательности ядерной локализации (NLS – nuclear localization signal). Химерная sgРНК – единая молекула РНК, вторичная структура которой имитирует комплекс зрелой crРНК и tracrРНК за счет структуры стебель-петля (повтор-антиповтор в комплексе crРНК-tracrРНК). С инженерной точки зрения, sgRNA состоит из двух частей: константной части, которая образуется из нескольких петель и связывает Cas9, и 5′-концевой короткой вариабельной спейсерной части, состоящей из 20 нуклеотидов. Последовательность спейсера обеспечивает специфичность узнавания ДНК-мишени, комплементарна участку генома, на котором CRISPR/Cas9 система произведет редакцию (протоспейсера) и определяется интересами исследователя, его целями и задачами. При этом есть небольшое ограничение в виде строго обязательного наличия, примыкающего к протоспейсеру на 3’‑конце, мотива PAM (NGG). Cas9 внесет разрез в область протоспейсера на расстоянии трех нуклеотидов перед мотивом PAM. Для транскрипции sgRNA необходима промоторная последовательность, чаще всего, в вектор включают промотор U6 РНК-полимеразы III. Кроме того, в плазмиды могут быть встроены дополнительные элементы, облегчающие селекцию несущих плазмиду клеток, например, оранжевый флюоресцирующий белок (OFP reporter) или гликопротеин CD4 [3,56].
После внесения системой CRISPR/Cas9 двухцепочечных разрывов репарация ДНК в клетке проходит либо по механизму негомологичного соединения концов, либо путем гомологичной рекомбинации. Негомологичное соединение концов как правило приводит к возникновению ошибок в последовательности, в результате чего в целевом локусе появляются мутации по типу инделей (инсерции, делеции). При гомологичной рекомбинации неповрежденный гомолог служит матрицей для восстановления исходной структуры ДНК. Это событие происходит в клетке довольно редко, однако использование адаптированной системы CRISPR/Cas9 позволяет повысить вероятность прохождения гомологичной рекомбинации на несколько порядков. Если добавить к компонентам CRISPR/Cas9 искусственно синтезированную молекулу ДНК, имеющую гомологию с последовательностью нуклеотидов в месте разрыва, то она может служить матрицей для другого способа репарации по механизму гомологичной рекомбинации, при котором небольшой фрагмент искусственной матрицы встраивается в целевой локус. В качестве такой матрицы чаще всего используют два типа конструкций: одноцепочечные олигонуклеотиды или плазмидные векторы. В первом случае искусственно синтезируют олигонуклеотиды, гомологичные сайту внесения двухцепочечного разрыва, оптимальная длина которых составляет около 90 нуклеотидов. Такие олигонуклеотиды могут содержать небольшие отличия от целевого сайта. При использовании плазмидных векторов в качестве донорских молекул для рекомбинации в них клонируют достаточно длинные плечи гомологии (от 500 до нескольких тысяч пар нуклеотидов). Такие плечи гомологии могут фланкировать дополнительные элементы, например, репортерные гены, гены устойчивости к антибиотикам и так далее. С помощью гомологичной рекомбинации в целевой локус можно поместить сайт рестрикции, маркерную метку или же нуклеотиды для исправления «неправильных» ДНК. Однако гомологичная рекомбинация активно происходит только в делящихся клетках, и ее эффективность очень сильно зависит от типа клетки, стадии жизнедеятельности, а также целевого локуса генома и самой матрицы [55,57].
Общая стратегия геномной инженерии с помощью системы CRISPR/Cas9 включает четыре основных этапа:
— выбор целевой нуклеотидной последовательности в геноме,
— создание нуклеазной конструкции, направленной на выбранную мишень,
— доставка конструкции в клеточное ядро,
— анализ полученных мутаций;
Выбор целевой нуклеотидной последовательности в геноме и тип используемой конструкции зависят от целей и задач экспериментатора.
С помощью системы CRISPR/Cas9 можно выполнить следующие типы редакции генома:
— негомологичное соединение концов в отсутствие донорной плазмиды опосредует делеции или инсерции нескольких нуклеотидов сайта-мишени и, как один из результатов, генный нокаут из-за мутаций рамки считывания и образования стоп-кодонов;
— в присутствии двухцепочечных олигонуклеотидов или донорной плазмиды фрагменты ДНК длиной свыше 14 т.п.н. могут встраиваться посредством лигирования, опосредованного негомологичным сшиванием концов;
— одновременное внесение нескольких двухцепочечных разрывов может приводить к делециям, инверсиям или транслокациям участков ДНК, расположенных между этими разрывами;
— гомологичная рекомбинация в присутствии донорной плазмиды с плечами гомологии, фланкирующими встраиваемый фрагмент, линейной донорной последовательности с гомологией менее 50 п.н. или олигонуклеотида приводит к внедрению одного или нескольких трансгенов для коррекции или замены существующих генов [58–61].
Редактирование генома возможно, как in vitro при доставке элементов систем CRISPR/Cas9 в культуры клеток, так и in vivo с помощью инъекций мРНК в зиготы.
Методы доставки векторов, экспрессирующих компоненты системы CRISPR/Cas9, в культивируемые клетки довольно разнообразны [48–50]. Наиболее распространенными и эффективными являются такие способы, как нуклеофекция, электропорация, липофекция, доставка, опосредованная вирусами. Для эффективной адресной доставки in vivo с помощью вирусов, наиболее перспективными являются векторы, неинтегрирующиеся в редактируемый геном, в частности векторы, основанные на аденовирусах, аденоассоциированных вирусах и некоторых лентивирусах. Для трансформации клеточных культур человека, мыши и других организмов чаще используют плазмидные векторы [65].
В случае трансформации целого организма разработан метод, основанный на микроинъекции мРНК сas9 и sgRNA в одноклеточные эмбрионы. Этот метод активно применяют у мышей, полосатого данио (Danio rerio) и дрозофилы. Для широкомасштабного охватывающего геном нокаута с использованием больших библиотек sgRNA используют лентивирусные векторы.
С точки зрения удобства и простоты использования, а также ожидаемой эффективности наибольшие перспективы для редакции генома овец имеет применение системы CRISPR/Cas9 направленное на внесение делеций или инсерций при запуске механизма негомологичного соединения концов для генного нокаута вызванного мутациями рамки считывания и образования стоп-кодонов. Таким образом могут быть отредактированы гены негативных регуляторов мышечного роста и развития, например, гена миостатина. «Отключив» синтез функционально активных белков, ограничивающих дифференцировку и пролиферацию мышечных клеток можно получить животных с фенотипом двойной мускулатуры, обладающих повышенной мясной продуктивностью. Преимуществом этого вида редакции является его относительная простота и ожидаемая высокая эффективность редакции, связанная с частотой срабатывания механизма негомологичного соединения концов [66]. К тому же для проведения такого геномного редактирования не требуются дополнительные матричные последовательности. Однако, такой тип редакции не позволит добиться оверэкспрессии генов, стимулирующих рост и развитие организма, а также «научить» животное производить модернизированные или несвойственные ему белки, что имеет особые перспективы для реализации в сельском хозяйстве и биотехнологических проектах [67–69].
Более сложным способом «заглушить» работу целевого гена является CRISPR-интерференция, основанная на использовании модифицированных белков Cas9, лишенных возможности разрезать цепи ДНК. Если Cas9 лишают одного нуклеазного домена, то белок становится никазой (nCas9) — режет только одну цепь ДНК, — а если лишают сразу двух, то белок становится инактивированным, или «мертвым» (dead, dCas9). Связывание комплекса направляющей РНК и модифицированного Cas9 с промотором какого-либо гена блокирует его взаимодействие с транскрипционными факторами и таким образом отключает его экспрессию. Модифицированную систему CRISPR-dCas9 можно также использовать для стимуляции транскрипции генов (активация спящих генов, оверэкспрессия) для чего к системе прикрепляют специальные активирующие домены. В целом систему CRISPR-dCas9 можно использовать для репрессии целых наборов генов или как платформу для конструирования более сложных регуляторных и модифицирующих комплексов. Для эпигенетической модификации нужных зон достаточно добавить модифицирующий домен. А пометив dCas9 флуоресцентными белками, можно визуализировать разные области хромосом.
Отредактировать последовательности существующих генов или внести неспецифичные для животной последовательности ДНК можно используя систему CRISPR/Cas9 с дополнительными донорными последовательностями. Так в присутствии двухцепочечных олигонуклеотидов или донорной плазмиды посредством лигирования, опосредованного негомологичным сшиванием концов в геном животного могут встраиваться фрагменты ДНК длиной свыше 14 т.п.н, а в присутствии донорной плазмиды с плечами гомологии, фланкирующими встраиваемый фрагмент можно добиться запуска процесса гомологичной рекомбинации и внедрения одного или нескольких трансгенов для направленной коррекции или замены существующих генов. Однако, широчайшие возможности такого метода, сопряжены с его высокой сложностью и крайне низкой эффективностью [70–72]. Для получения гомозиготных мутантов, передающих приобретённые в ходе CRISPR/Cas редакции мутации по наследству разработан метод мутагенной цепной реакции, основанный на встраивании в геном эукариот работоспособной воспроизводящейся CRISPR/Cas9 кассеты.
Перспективным является подход «редактирования оснований» с использованием системы CRISPR/Cas9. Большинство методов, разработанных на основе системы CRISPR-Cas, предполагают разрезание обеих цепей геномной ДНК с последующей починкой двойных разрывов, что чревато ошибками. Альтернативный подход — «редактирование оснований» (base editing) — состоит в химической модификации нуклеотидов без создания двойных разрывов. Разработан метод превращения пар C•G в T•A при помощи фермента цитозиндезаминазы, присоединенного к модифицированному белку Cas9. Для этого используют мутантный вариант Cas9 с никазной активностью (никаза — фермент, разрезающий только одну из двух нитей двойной спирали). На этой основе был разработан метод «редактирования оснований» (base editing), позволяющий аккуратно заменять комплементарную пару оснований C•G на T•A. Для этого к мутантному Cas9 при помощи «мостика» из нескольких аминокислотных остатков пришивают фермент цитидиндезаминазу. Этот фермент превращает цитозин (C) в урацил (U), который при транскрипции и репликации «читается» ферментами-полимеразами как тимин (T). Вторая, комплементарная нить ДНК надрезается белком Cas9, и в ходе починки (репарации) этой нити напротив урацила ставится аденин (А). Техническая трудность состоит в том, что урацила в норме не должно быть в составе ДНК. Ферменты репарации внимательно следят за тем, чтобы никаких урацилов в ДНК не было, а в случае их обнаружения пытаются вырезать «бракованный» фрагмент и выстроить его заново на матрице комплементарной цепи ДНК. Для того, чтобы система «редактирования оснований» исправно работала, к ней приделывают еще один компонент — ингибитор фермента, вырезающего урацилы из ДНК.
4 Подготовка материально-технической базы для проведения экспериментов по редактированию генома
Эксперимент был выполнен на овцах северо-кавказкой мясо-шерстной породы в возрасте 1,5-2 года, со средней массой 48-54 кг. Биопсия кожи была взята из дермального слоя кожи у основания ушной раковины животного. Все животные были клинически здоровыми и содержались в одинаковых условиях вивария факультета ветеринарной медицины и технологического менеджмента.
Для получения фибробластов и дальнейшей работы с клеточными культурами использовали:
Ламинарный шкаф LABGARD NU-425-400E II класса биологической безопасности для защиты оператора, продукта и окружающей среды, с рециркуляцией воздуха, производства компании «NuAire», США (Рисунок 10). Ламинарный шкаф имеет следующие особенности:
- Максимум защиты персонала, продукта и окружающей среды;
- Равномерный ламинарный поток воздуха с полным отсутствием турбулентности воздушного потока, поступающего на рабочую поверхность;
- Система Hepex исключает утечку загрязненного воздуха: пленум окружен зонами негативного давления.
- Полностью стальной корпус;
- Большая съемная стальная рабочая поверхность;
- Вертикально скользящее безопасное стекло, поднимающееся вручную, со стандартным доступом в рабочую зону 20 см. В нерабочем состоянии стекло полностью опускается, закрывая рабочую зону;
- Лёгкость в управлении и техническом обслуживании;
- Эргономичный дизайн;
- Гибкость конфигурации;
- Соответствие международным стандартам.
Технические характеристики ламинарного бокса представлены в таблице 2.
Таблица 2 – Технические характеристики ламинарного бокса.
| Параметры NU-425-400E | |
| Внешние размеры, В х Ш х Г (мм) | 1600х1362х835 |
| Размеры рабочей зоны, В х Ш х Г (мм) | 724х1178х597 |
| Электропитание, В/Гц | 220/50 |
| Статическое давление, мм водного столба | 1.27 – 2.54 |
| Скорость защитного потока воздуха (м/с) | 0,53 |
Общие характеристики ламинарного шкаф NU-425-400E следующие: надёжная конструкция, предназначенная для долговечной и бесшумной работы; модель LabGard NU-425 имеет полностью сварной, полированный корпус из 100% нержавеющей стали; каркас шкафа специально укреплен для уменьшения вибрации, уровня шума и поддержания плоских поверхностей; стальные поверхности долговечны и легко подвергаются обработке дезинфектантами; все воздуховоды, пленум, технические отверстия герметично закупорены для предотвращения утечек и контаминации в лаборатории.
Контрольный блок модели, расположенный на уровне глаз, содержит выключатели и индикаторы, такие как:
- включение/выключение флуоресцентного и ультрафиолетового освещения;
- включение/выключение двигателя вентилятора, нагнетающего поток воздуха;
- включение/выключение внутренних электрических розеток;
- манометр, показывающий нагрузку на Hepa фильтр;
- включение/выключение аварийного акустического сигнала при низком/высоком уровне стекла (индикатор тревоги);
- регулятор скорости потока воздуха (только для сервиса).
Стандартная спецификация ламинарного бокса следующая: Hepex система, обеспечивающая отсутствие утечки воздуха из рабочей камеры; Hepa фильтр с эффективностью 99,9% для частиц размером 0,3 микр.; внешнее флуоресцентное освещение рабочей поверхности; возможность смены фильтра через переднюю панель; вертикально скользящее стекло; съёмная рабочая поверхность; металлическая решётка, защищающая основной фильтр; закалённое стекло; съёмный контрольный центр; дренажное устройство; манометр; две электрические розетки; один сервисный кран; одно сервисное отверстие.

Рисунок 10 – Ламинарный шкаф.
Высокоскоростная центрифуга «СМ-50» для пробирок Eppendorf производитель компания ELMI (Латвия). Количество пробирок в роторе составляет 12 штук. Центрифуга СМ-50 является одной из самых компактных и мощных мини-центрифуг. Благодаря запатентованной аэродинамической камере и магнитной крышке ротора, шум и нагрев снижены до низких величин, что создает новый стандарт в центрифугировании. Описание параметров центрифуги «СМ-50» представлены в таблице 3.
Таблица 3 – Основные параметры центрифуги «СМ-50».
| Параметры центрифуги «СМ-50» | |
| Скорость вращения ротора, об/мин | 1000-16000 (дискретность 1000) |
| Гравитационное поле, G max | 18000 (дискретность 1000) |
| Максимальный шум, Дб | 46 |
| Угол наклона пробирок в роторе, град. | 45 |
| Система торможения | трёхступенчатая, отключаемая |
| Минимальное время торможения (с 16000 об/мин), сек | 12 |
| Питание от сети, В/Гц | 220/50 |
| Количество пробирок в роторе | 12 |
| Объемы применяемых пробирок | 0,5; 1; 1,5; 2 |
| Потребляемая мощность, Вт | 200 |
| Габаритные размеры (Ш х Д х В), мм | 256х218х175 |
Твердотельный термостат «Bio TDB-100» с функцией охлаждения и нагрева производитель компания BioSan (Латвия). Цифровой термостат Bio TDB-100 предназначен для использования при температуре окружающей среды от +4 до +40 С и высокой влажности до 80%, может быть использован в инкубаторах. Прибор в стандартной комплектации включает алюминиевый блок на 24 лунки для пробирок объемом 2-1,5 мл, 15 лунок для пробирок объемом 0,5 мл и 10 лунок для пробирок объемом 0,2 мл. Характеристики термостата «Bio TDB-100»: температурный диапазон в интервале от +25 до +100 C°; шаг задания температуры 0,1°C; равномерность распределения температуры при +37 ± 0,1°С; установлена защита от перегрева; цифровой таймер от 1 минуты до 96 часов или непрерывно; диаметр блока130 мм; глубина блока 45 мм; габариты 210 x 230 x 115 мм.
Весы аналитические GR-202, производитель: Япония (Рисунок 11). При помощи GR-202 можно легко производить высокоточное взвешивание в микродиапазоне, дискретность которого при этом будет равна 0,01 мг. Все операции, касающиеся взвешивания, производят довольно быстро. К преимуществу аналитических весов можно также отнести простоту их эксплуатации и долговечность, а также повышенную износостойкость платформ у большинства подобных весов. Основные функции: выбор единиц измерения (грамм, карат, фунт, унция и т.д.); режимы процентного взвешивания и штучного подсчета с функцией ACAI; возможность измерения плотности веществ и работы с магнитным материалом при помощи стандартного поддонного крюка; возможность автономной работы с помощью встраиваемого аккумулятора (опция); соответствия нормам GLP; функция автоматического включения/выключения.

Рисунок 11 – Аналитические весы.
Для выращивания и исследования клеточных культур применяли лабораторный инкубатор с атмосферой СО2 (Рисунок 12). Благодаря системе прямого обогрева отпадает необходимость в применении вентилятора, исключаются риски возникновения вибрации и взаимного загрязнения проб. Инфракрасный датчик измеряет концентрацию СО2 без отклонений, обеспечивает максимальную надёжность и точность измерений в течение всего процесса. Благодаря уникальной системе прямого обогрева камеры существенно упрощается установка и техническое обслуживание аппарата. Внутренняя стеклянная дверь уплотнена на стыке с изоляцией камеры, что позволяет контролировать пробы без нарушения условий внутренней среды. Наружная дверь уплотнена собственной наружной прокладкой.

Рисунок 12 – Лабораторный инкубатор с атмосферой СО2
Инкубатор СО2 имеет такие плюсы, такие как: обеззараживание при 160 °C или стерилизация при 200 °C, при этом датчик CO2 остаётся внутри аппарата; противомикробное покрытие; внутренняя стеклянная дверь; опасность загрязнения предотвращается благодаря отсутствию вентилятора; 2 x 3 светодиодный дисплей; непрерывная индикация текущих значений температуры и концентрации СО2; аудиовизуальная сигнализация тревоги; инфракрасный датчик СО2 без погрешности измерения; независимый защитный термостат; Hepa-фильтр на подводящем трубопроводе СО2; бесшовная внутренняя камера с округлёнными углами; шестисекционная система прямого обогрева, обеспечивающая максимальную однородность внутренних условий проходной изолятор 25 мм на задней стороне аппарата. Лабораторный инкубатор с атмосферой СО2 имеет параметры:
- Внутренний объём: 50, 190 литров;
- Рабочая температура: от 5 °C выше температуры окружающей среды
до 60 °C;
- Неуправляемая относительная влажность: до 95% RH при 37 °C;
- Концентрация CO2: 0,2 % – 20 %;
- Датчик CO2: инфракрасный датчик (IR), обеспечивающий измерения без отклонений;
- Внутренняя камера: исполнение Standard – нержавеющая сталь.
Сетчатые полки можно загружать не больше, чем приблизительно до 50% площади, и по мере возможности так, чтобы обеспечивалось равномерное движение воздуха во внутреннем пространстве камеры.
Для работы с компетентными клетками использовался Шейкер‑инкубатор ES-20/60, производитель компания BioSan (Рисунок 13). Прибор обеспечивает плавное или интенсивное перемешивание в колбах, установленных на платформе. Имеет особенности: диапазон регулирования скорости перемешивания: 50 – 250 об/мин; высокоточное распределение температуры по всему объему камеры шейкера-инкубатора (от комнатной температуры до +80°С); внутренняя камера выполнена из нержавеющей стали; низкое энергопотребление, несмотря на относительно большие размеры; Технические характеристики прибора представлены в таблице 4.
Таблица 4 – Технические характеристики шейкера-инкубатора
| Шейкер-инкубатор ES-20/60 | |
| Диапазон регулирования скорости, об/мин | 50 — 250 (шаг 10 об/мин) |
| Диапазон установки температуры, °C | от 25 до 80 |
| Диапазон регулирования температуры, °C | от 10 выше комнатной до 80 |
| Шаг установки температуры, °C | 0,1 |
| Стабильность температуры, °C | ±0,5 |
| Максимальная нагрузка, кг | 8 |
| Орбита, мм | 20 |
| Цифровая установка времени | 1 мин — 96 ч / непрерывно
(шаг 1 мин) |
| Дисплей ЖК, | 2 × 16 знаков |
| Максимальное время непрерывной работы | до 30 суток |
| Размеры (Д×Ш×В), мм | 590 × 525 × 510 |
| Размеры рабочей камеры, мм | 460 × 400 × 310 |

Рисунок 13 – Шейкер-инкубатор
Для учета результатов исследований использовали инвертированный микроскоп Olympus IX71 (Рисунок 14). Для микроскопа характерны высочайшие оптические качества, обеспеченные оптикой UIS мирового класса, надежность и простота в обращении. Микроскоп укомплектован микроманипуляторами, нагреваемым столиком, оптикой Хоффмана модуляционного контраста. Используемый микроскоп обеспечивает высококачественную фотодокументацию и видеосъемку. Простые дополнения к системе позволяют использовать микрофотометрию, видеозапись и трехмерные наблюдения.
 Рисунок 14 – Инвертированный микроскоп Olympus IX71.
Рисунок 14 – Инвертированный микроскоп Olympus IX71.
В дополнение к наблюдательному порту, порту бокового обзора и порту DSLR-камеры, входящим в основную конфигурацию IX71, могут быть добавлены право/лево — сторонний порт и порт для видеокамеры. Конфигурация системы, таким образом, становится максимально гибкой. Прямая передача свободного от аберраций изображения. Вдоль оптической оси объектива на IX71 может быть установлена CCD-камера для прямой записи свободного от аберраций первичного изображения, сформированного объективами UIS2. Такое прямое изображение передает полностью прекрасное оптическое качество системы на сенсоры измерительного устройства без каких-либо дополнительных оптических элементов. Эта возможность уникальна для Olympus IX71 и идеальна для приложений, требующих высочайшего разрешения и качества. Встроенный промежуточный регулятор изменения увеличения. IX71 снабжен регулятором изменения увеличения с 1х и 1,6х промежуточными коэффициентами, что позволяет быстро менять увеличение простым движением одной руки. Фото- и видео- документирование. На микроскоп Olympus IX71 могут быть установлены любые устройства для документирования: цифровая фото- или видеокамера.
Для наблюдения за трансфицированными клетками использовали микроскоп Olympus BX41, дополненный блоком флуоресценции (Рисунок 15). Исследовательский микроскоп разработан для проведения комплексных экспериментов и исследований в разных областях. Имеет небольшие размеры. Позволяет использовать следующие методы обзора: светлое поле; темное поле; фазовый контраст; поляризованный свет; флуоресценция. Все ручки управления (включение/выключение, регулировка накала лампы, фокусировка, перемещение препарата) сведены в единую рабочую зону. Замок фокусировки для сохранения положения фокуса при смене образцов. Возможность комплектации микроскопа блоком флуоресценции. Флуоресцентные изображения обладают высокой яркостью и четкостью. Флуоресцентный осветитель с ртутной лампой мощностью 50 Вт снабжен центрируемой полевой диафрагмой, портом для нейтральных светофильтров и двухпозиционным слайдером для фильтровых кубов. На выбор поставляются кубы УФ, синего, зеленого или желтого освещения. Микропроцессорный блок поджига лампы включает автоматическую систему питания лампы с памятью подобранного режима, а также LCD счетчик продолжительности работы. Осветитель проходящего света, реализующий принцип Келера; галогеновая лампа 6В 30Вт. Опционально – светодиодный источник света или плоско-вогнутое зеркало. Фокусировка достигается вертикальным перемещением столика с помощью роликового механизма (рейка и зубчатое колесо). Грубый ход при перемещении: 36.8 мм, точный ход при перемещении: 0,2 мм, полный ход: 25 мм. Регулировка натяжения перемещения ручки. Ограничитель верхнего предела подъема столика.

Рисунок 15 – Микроскоп Olympus BX41.
Для выделения плазмидной ДНК (вектор 111, вектор 222) набором Plasmid Midiprep 2.0, Евроген использовали настольную высокоскоростную лабораторную центрифугу SIGMA 3K30 (Sigma Laborzentrifugenс, Германия), с ускорением свыше 60 000 g, диапазон установки температуры –20…+40°C (Рисунок 16). Центрифуга содержит большой выбор роторов, максимальная ёмкость 8×85 мл, максимальная скорость 30 000 об/мин, включены 50 программ, имеется таймер.

Рисунок 16 – Центрифуга SIGMA 3K30
Центрифуга содержит функцию предварительного охлаждения ротора, что важно в ходе нашего исследования. Достоинства управления SIGMA 3K30: установка параметров центрифугирования с помощью одной поворотной кнопки; микропроцессорный контроль параметров центрифугирования; большой графический дисплей, отражающий одновременно скорость RPM, ускорение RCF и время; непрерывная работа, таймер 10 сек – 9 ч 59 мин; 50 программ центрифугирования со свободным программированием всех параметров; 10 линейных кривых ускорения и торможения; система автоматического распознавания ротора, ограничивающая установку значений скорости до максимально разрешенных для каждого ротора; моторизованная блокировка крышки; индукционный (бесщеточный) двигатель, не требующий сложного обслуживания; минимальный уровень шума при максимальной скорости 64 дБ. Технические характеристики представлены в таблице 5.
Таблица 5 – Технические характеристики высокоскоростной лабораторной центрифуги SIGMA 3K30.
| Sigma Laborzentrifugenс | |
| Максимальная вместимость, мл | 6 x 85 |
| Минимальная скорость, об/мин | 100 |
| Максимальная скорость, об/мин | 30 000 |
| Максимальное ускорение, сек (ротор 11190) | 30 |
| Максимальное ускорение, сек (ротор 12158) | 90 |
| Максимальное торможение, сек (ротор 11190) | 14 |
| Максимальное торможение, сек (ротор 12158) | 58 |
| Уровень шума при максимальной скорости, дБ, не более | 64 |
| Напряжение сети | 220-240 В,50/60 Гц |
| Потребляемая мощность, Вт | 1260 |
| Размеры (В х Ш х Г), мм | 400 х 550 х 650 |
Мешалка Vortex ZX3 (VELP, Италия), предназначена для перемешивания различных веществ в пробирках с помощью встряхиваний и круговых движений (Рисунок 17). Вортекс ZX3 используется для длительного непрерывного перемешивания и для перемешивания с меняющейся частотой вибрации. Мешалка изготовлена из алюминия и покрыта эпоксидкой, что дает защиту от химических и механических повреждений, а также от коррозии в целом. Имеет свои характеристики: диаметр крепления датчика 55 мм; частота перемешивания от 0 до 40 Гц; мощность составляет 45 Вт; габариты – 150 х 150 х 134 мм; электропитание – 110-120 В, 60 Гц; 220-240 В, 50 Гц; 220-240 В, 60 Гц. В нашей работе вортекс использовался на этапе выделения плазмидной ДНК (Crispr Ovis aries) набором Plasmid Midiprep 2.0, Евроген. С помощью вортекса перемешивали суспензию, включающую ДНК, растворы (нейтрализующий, лизирующий, промывочный, элюирующий, раствор для удаления эндотоксинов).
 Рисунок 17 – Вортекс ZX3
Рисунок 17 – Вортекс ZX3
Выделение ДНК из биологического материала (кровь, культура клеток) проводилось в специальном ламинарном боксе производитель компания LAMSYSTEMS (Рисунок 18). Ламинарный бокс имеет отличительные особенности: механизм подъема фронтального стекла позволяет фиксировать его в двух положениях, что облегчает обработку дезрастворами рабочей камеры, не имеющей крепежных элементов и выступов, а также самого стекла с обеих сторон; наклон передней панели улучшает обзор рабочей камеры и увеличивает полезную площадь столешницы; все системы электроуправления расположены за пределами основного корпуса бокса для легкости доступа; время УФ-обработки рабочей камеры бокса устанавливается с помощью таймера, отображающего также и время общей наработки лампы блока УФО; корпус ламинара окрашен порошковой эмалью, стойкой к обработке дезрастворами; рабочая столешница выполнена из нержавеющей стали; на подставке предусмотрены винтовые опоры для фиксации и регулировки положения бокса и колесные опоры, предназначенные исключительно для удобства перемещения при установке на место эксплуатации; микропроцессорная система управления двигателем вентилятора без применения энергопреобразующих силовых элементов – SintelL 1. Система позволяет максимально снизить уровень электропотребления работающего бокса, уменьшить уровень акустических шумов и помех; система статической стабилизации воздуха AIS LS обеспечивает постоянный воздушный баланс внутри рабочей камеры ламинарного бокса вне зависимости от степени загрязненности фильтра; блок освещения вынесен за пределы рабочей камеры ламинара и не вызывает турбулентности потока воздуха; панель управления с ЖК-дисплеем индицирует включение систем изделия, их возможные неисправности, выбранные режимы работы и технологический таймер; фильтр Нepa поджат с помощью пружин, обеспечивающих герметичность уплотнения фильтра на весь срок эксплуатации; электронная панель управления обеспечивает легкость эксплуатации и дезобработки. Применяли низкоскоростную центрифугу-вортекс «FVL-2400N Combi-spin» производитель компания BioSan. Скорость вращения вортекса — 2800 оборотов в минуту. В комплект входят два ротора: на 12 пробирок объемом 1,5 мл; ротор на 12 пробирок объемом 0,5 – 0,2 мл. FVL-2400N имеет на одном модуле возможность перемещения и разделения образцов. В работе использовали мини-ротатор для пробирок и вакутайнеров Bio RS-24 производитель компания BioSan. Прибор осуществляет вертикальное вращение платформы. Ротатор идеальный инструмент для предотвращения свертывания крови в пробирках, проведения процессов экстракции биологических компонентов. Может эксплуатироваться в холодных комнатах и биологических инкубаторах при температуре от +4°С до +40°С. Спецификация прибора: диапазон регулирования скорости 5‑30 об/мин; цифровая установка времени 1 минута – 24 часа / непрерывно; вертикальное вращение 360°; максимальное время непрерывной работы 8 часов; размеры (длина 325 мм, ширина 190 мм, высота 155 мм); потребляемый ток / мощность – 12 В, 110 мA/1,3 Вт. Высокоскоростная центрифуга «СМ-50» производитель компания ELMI (Латвия), описанная ранее, также использовалась на данном этапе. Для охлаждения и нагрева пробирок с растворами применяли твердотельный термостат «CH-100» производитель компания BioSan (Латвия). Основные параметры прибора «CH 100»: диапазон установки температуры от ‑10°C до +100°C; диапазон регулирования температуры 30°C ниже комнатной +100°C; шаг установки температуры 0,1°C; стабильность температуры ±0.1°C; равномерность распределения температуры при 37°C±0,1°C; цифровая установка времени 1 минута – 96 часа; дисплей ЖК,2× 16 знаков; потребляемый ток / мощность – 12 В, 4,4 A / 55 Вт.
Для определения концентрации ДНК использовали прибор Nanophotometer «ImpLEN» (Рисунок 19). Встроенный вортекс обеспечивает однородность образца с наивысшей степенью точности с наименьшим объемом образца всего 0,3 мкл. Полные возможности сканирования варьируются от 200 — 900 нм для быстрого и полного анализа образцов за 3,5 сек.
Прибор имеет свои характеристики: диапазон обнаружения dsDNA 1 нг / мкл до 16 500 нг / мкл; минимальный размер выборки 0,3 мкл; фотометрический диапазон (эквивалент 10 мм) от 0 до 330 А; воспроизводимость длины волны ± 1 нм; точность длины волны ± 0,75 нм; пропускная способность1,8 нм; лампа Ксеноновая рассчитана на 10 лет эксплуатации; Совместимость программного обеспечения Windows 7, 8, 10 (32 и 64 бит), OS X, iOS и Android OS; вортекс 2800 об / мин; размер трубки до 2,0 мл
Рисунок 18 – Ламинарный бокс. 

Рисунок 19 – Nanophotometer
Для приготовления ПЦР смеси использовали ПЦР-бокс «Cleaner ПЦР UVC/T-M-AR» производитель компания BioSan (Рисунок 20). Бокс оснащен одной открытой УФ‑лампой, установленной в верхней части бокса. УФ‑излучение дезинфицирует рабочую поверхность, инактивируют фрагменты ДНК/РНК в течение 15–30 минут. Цифровой таймер контролирует длительность прямого ультрафиолетового облучения. Лампа дневного света обеспечивает освещение рабочего места. Бокс оснащен бактерицидным проточным УФ-рециркулятором воздуха AR, обеспечивающим постоянную дезинфекцию внутри бокса во время работы с ДНК/РНК ампликонами. УФ‑рециркулятор воздуха AR состоит из УФ‑лампы, вентилятора и антипылевого фильтра, заключенных в специальный корпус. Включенный рециркулятор увеличивает максимум плотности УФ-лучей, что является достаточно эффективным для ДНК/РНК инактивации. Преимущества боксов Biosan: УФ деконтаминация высокой плотности без озона; автоматическое выключение УФ ламп в случае открытия передней дверцы; бактерицидный проточный рециркулятор, обеспечивающий постоянное обеззараживание внутреннего пространства бокса во время работы; стенки из ударопрочного  стекла; низкий уровень шума и энергопотреблении.
стекла; низкий уровень шума и энергопотреблении.
Рисунок 20 – ПЦР-бокс
Для амплификации целевых участков в гене MSTN использовали амплификатор Терцик «ДНК-технология». Прибор содержит 4 независимых термоблока, прост в настройках. Присутствует дисплей для отображения всех действий. Термоблок поддерживает равномерную температуру внутри из‑за зеркальности расположения матриц и пробирок на 0,5 мл в термоблоке. Многоканальность прибора позволяет: одновременно ставить несколько различных диагностикумов при комплексных исследованиях; независимо работать нескольким пользователям; ускорить оптимизацию новых реакций; обрабатывать до 40 образцов за один цикл работы прибора, по одной программе при массовых исследованиях. Также применялся горизонтальный форез нуклеиновых кислот в агарозном геле с помощью источника питания PowerPac Basic «BIO-RAD» (Рисунок 21). Базовый прибор PowerPac Basic работает при установленном напряжении с прецизионной точностью поддержания параметров для высокой результативности.
 Чтобы предотвратить повреждение электрофоретической ячейки, PowerPac Basic обеспечивает автоматическое переключение на постоянный ток или постоянное напряжение в зависимости от того, что достигается раньше. Яркий LED-экран выводит любой из параметров — ток, напряжение, время. Возможно одновременное подключение до 4 электрофоретических камер.
Чтобы предотвратить повреждение электрофоретической ячейки, PowerPac Basic обеспечивает автоматическое переключение на постоянный ток или постоянное напряжение в зависимости от того, что достигается раньше. Яркий LED-экран выводит любой из параметров — ток, напряжение, время. Возможно одновременное подключение до 4 электрофоретических камер.
Рисунок 21 – Источник питания, камера для горизонтального фореза.
Результаты гельэлектрофореза фрагментов ДНК, определяли с помощью системы гель-документирования ChemiDoc XRS+ производитель компания Bio-Rad (Рисунок 22). Система гель-документирования Gel Doc XR+ создана на базе CCD камеры высокого разрешения и чувствительности, и позволяет решать основные задачи анализа и документирования гелей и мембран. Gel Doc XR+ подходит для работы с широким спектром образцов, от полиакриламидных и агарозных гелей до блотов, и позволяет использовать такие методы детекции, как денситометрия, флуоресценция и колориметрия. Благодаря полной автоматизации всех процессов и программного обеспечения Image Lab результаты получаются точные и воспроизводимые. Технические характеристики прибора представлены в таблице 6.
Рабочая комплектация системы: CCD камера; трансиллюминатор (УФ и видимый свет); защитный колпак («темная комната»); комплект кабелей для коммутации; оригинальное ПО Image Lab.
Таблица 6 – Технические характеристики системы гель-документирования ChemiDoc XRS+.
| Камера CCD, | 12 бит |
| Разрешение камеры, МПи | 4 |
| Динамический диапазон | боле 3-х порядков |
| Источник света трансиллюминатор | (белый свет, УФ 302 нм) |
| Эмиссионные фильтры | EtBR/SYBR Green BP (опционально до 6-ти красителей) |
| Максимальный размер геля, см | 28 х 36 |
| Максимальный размер сканирующей области, см | 19,4 х 26 |
| Габариты, мм | 600 х 360 х 960 |

Рисунок 22 – Система гель-документирования Gel Doc XR+
В ходе эксперимента использовались стерильные пробирки (1,5 мл; 5 мл; 15 мл; 45 мл), стерильная посуда, наконечники разных объемов, пипетки (от 1 мкл до 5 мл), чашки Петри (одноразовые, стерильные), культуральные матрасы и дополнительные материалы.
5 Компьютерный анализ и конструирование гайдовых последовательностей РНК
В качестве целевого гена для редактирования с использованием системы CRISPR/Cas9 выбран ген миостатина, кодирующий один из ключевых регуляторов мышечного роста и развития. При блокировании действия миостатина наблюдается увеличение мышечной массы и повышение силовых характеристик скелетных мышц [73–75].
У овец ген миостатина расположен на второй хромосоме, имеет 3 кодирующих экзона и два интрона. Общая длина гена 2894 пар оснований. Длина белкового продукта MSTN после сплайсинга составляет 375 аминокислот (Рисунок 22). В С-концевой области (281-375 амк.) расположен функционально важный специфичный домен, определяющий принадлежность гена MSTN к семейству ростовых факторов Transforming growth factor β-family (TGF-β). В N-концевой части аминокислоты с 39 по 249 определяют последовательность гомодимера пропептида MSTN, который в дальнейшем взаимодействует с TGF-бета-связывающими белками и обеспечивает функциональную активность гена [76].
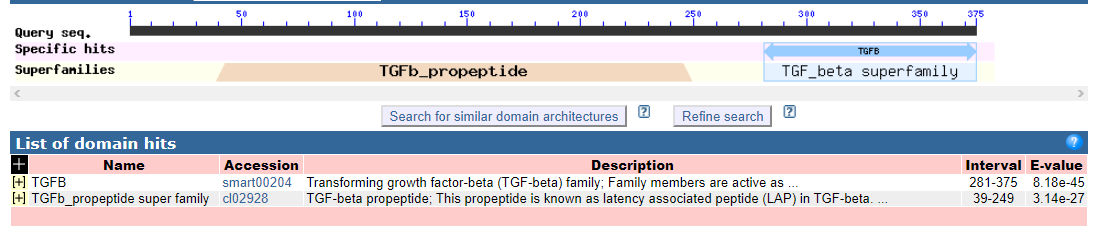
Рисунок 23 – Функционально значимые домены белка MSTN
Таким образом для нокаута гена миостатина необходимо добиться нарушения формирования функциональных доменов. Мутации можно вносить непосредственно в области, кодирующие эти домены, однако в таком случае высока вероятность того, что белок частично сохранит свои функции. В связи с этим целесообразно редактировать последовательности, стоящие «выше по течению» (upstream). Добившись сдвига рамки считывания в начальных областях гена можно быть уверенным, что мутантный белковый продукт будет весьма сильно отличаться от канонического и с низкой долей вероятности сможет выполнять его функции. В связи с этим было принято решение провести направленное редактирование кодирующей области первого экзона гена миостатина.
Выбор целевой последовательности и последующие конструирование одноцепочечных олигонуклеотидов имеет критически важное значение и во многом определяет успешность эксперимента. Последовательность-мишень должна иметь длинну от 19 до 20 нуклеотидов вместе с мотивом (PAM) NGG-протоспейсера на 3 ‘конце. Необходимо убедится, что целевая последовательность не содержит участков, гомологичных с другими генами, так как это может увеличить нецелевые эффекты. По опубликованных данным, комплекс gRNA-Cas9 потенциально может сработать даже при несоответствии в 1–3 нуклеотида. Можно выбрать целевую последовательность, кодирующую смысловую последовательность локус-мишень или антисмысловую последовательность. Таким образом, можно генерировать CRISPR РНК в двух возможные ориентации при условии, что они соответствует требованиям PAM на 3’ концах [77] .
В ходе анализа последовательности первого экзона гена миостатина установлено, что в его кодирующей области существует 30 потенциальных PAM мотивов (NGG): 14 на прямой цепи и 16 на обратной (Таблица 7).
Таблица 7 – Мотивы PAM в экзоне 1 гена миостатина
| Прямая цепь(strand+) 5’ — 3’ |
| ATGCAAAAACTGCAAATCTTTGTTTATATTTACCTATTTATGCTGCTTGTTGCTGGCCCAGTGGATCTGAATGAGAACAGCGAGCAGAAGGAAAATGTGGAAAAAAAGGGGCTGTGTAATGCATGCTTGTGGAGACAAAACAATAAATCCTCAAGACTAGAAGCCATAAAAATCCAAATCCTCAGTAAGCTTCGCCTGGAAACAGCTCCTAACATCAGCAAAGATGCTATAAGACAACTTTTGCCCAAGGCTCCTCCACTCCGGGAACTGATTGATCAGTACGATGTCCAGAGAGATGACAGCAGCGACGGCTCCTTGGAAGACGATGACTACCACGTTACGACGGAAACGGTCATTACCATGCCCACGGAGT |
| Обратная цепь (strand-) 5’ — 3’ |
| ACTCCGTGGGCATGGTAATGACCGTTTCCGTCGTAACGTGGTAGTCATCGTCTTCCAAGGAGCCGTCGCTGCTGTCATCTCTCTGGACATCGTACTGATCAATCAGTTCCCGGAGTGGAGGAGCCTTGGGCAAAAGTTGTCTTATAGCATCTTTGCTGATGTTAGGAGCTGTTTCCAGGCGAAGCTTACTGAGGATTTGGATTTTTATGGCTTCTAGTCTTGAGGATTTATTGTTTTGTCTCCACAAGCATGCATTACACAGCCCCTTTTTTTCCACATTTTCCTTCTGCTCGCTGTTCTCATTCAGATCCACTGGGCCAGCAACAAGCAGCATAAATAGGTAAATATAAACAAAGATTTGCAGTTTTTGCAT |
Примечание: Мотивы PAM выделены красным шрифтом
По результатам анализа литературных данных установлено, что зарубежными учеными ранее были проведены эксперименты по редактированию первого экзона гена миостатина овец с использованием шести различных целевых последовательностей (Таблица 8).
Таблица 8 – Варианты целевых последовательностей, используемые зарубежными учеными
| Y. Zhang, 2018 | ATGCAAAAACTGCAAATCTTTGTTTATATTTACCTATTTATGCTGCTTGTTGCTGGCCCAGTGGATCTGAATGAGAACAGCGAGCAGAAGGAAAATGTGGAAAAAAAGGGGCTGTGTAATGCATGCTTGTGGAGACAAAACAATAAATCCTCAAGACTAGAAGCCATAAAAATCCAAATCCTCAGTAAGCTTCGCCTGGAAACAGCTCCTAACATCAGCAAAGATGCTATAAGACAACTTTTGCCCAAGGCTCCTCCACTCCGGGAACTGATTGATCAGTACGATGTCCAGAGAGATGACAGCAGCGACGGCTCCTTGGAAGACGATGACTACCACGTTACGACGGAAACGGTCATTACCATGCCCACGGAGT |
| M. Wu, 2018 | ATGCAAAAACTGCAAATCTTTGTTTATATTTACCTATTTATGCTGCTTGTTGCTGGCCCAGTGGATCTGAATGAGAACAGCGAGCAGAAGGAAAATGTGGAAAAAAAGGGGCTGTGTAATGCATGCTTGTGGAGACAAAACAATAAATCCTCAAGACTAGAAGCCATAAAAATCCAAATCCTCAGTAAGCTTCGCCTGGAAACAGCTCCTAACATCAGCAAAGATGCTATAAGACAACTTTTGCCCAAGGCTCCTCCACTCCGGGAACTGATTGATCAGTACGATGTCCAGAGAGATGACAGCAGCGACGGCTCCTTGGAAGACGATGACTACCACGTTACGACGGAAACGGTCATTACCATGCCCACGGAGT |
| M. Crispo, 2015 | ATGCAAAAACTGCAAATCTTTGTTTATATTTACCTATTTATGCTGCTTGTTGCTGGCCCAGTGGATCTGAATGAGAACAGCGAGCAGAAGGAAAATGTGGAAAAAAAGGGGCTGTGTAATGCATGCTTGTGGAGACAAAACAATAAATCCTCAAGACTAGAAGCCATAAAAATCAAATCCTCAGTAAGCTTCGCCTGGAAACAGCTCCTAACATCAGCAAAGATGCTATAAGACAACTTTTGCCCAAGGCTCCTCCACTCCGGGAACTGATTGATCAGTACGATGTCCAGAGAGATGACAGCAGCGACGGCTCCTTGGAAGACGATGACTACCACGTTACGACGGAAACGGTCATTACCATGCCCACGGAGT |
Примечание: Мотивы PAM выделены красным шрифтом, целевые последовательности по прямой цепи выделены салатовым цветом, целевые последовательности по обратной цепи-синим цветом.
Для того чтобы понимать, на какую область аминокислотной последовательности приходятся используемые в экспериментах целевые нуклеотидные последовательности были определены их координаты относительно точки с.1-первого нуклеотида стартового кодона гена миостатина (Таблица 9).
Две из ранее используемых экспериментах целевых последовательностей направляют комплекс CRISPR/Cas9 в область кодирования аминокислотной последовательности пропептида, две в его пограничную область и две в вышестоящую область (последовательность пропетидного домена начинается с 39 аминокислоты, соответственно последовательность кодирующая входящие в него аминокислоты начинается с нуклеотида в позиции с.117).
Таблица 9 – Координаты целевых последовательностей, используемых зарубежными учеными для редактирования первого экзона гена миостатина у овец
| Редактирование экзона I у овец | |||||
| Год | Автор | Целевая последовательность 5’-3’ | PAM | Цепь | Координаты целевой последовательности в гене (отсчет от с.1) |
| 2018 | Zhang
[9] |
ATTTATGCTGCTTGTTGC | TGG | Прямая | 36-53 |
| TAAGACAACTTTTGCCCA | AGG | Прямая | 230-247 | ||
| 2018 | Wu
[10] |
ATTTATGCTGCTTGTTGC | TGG | Прямая | 36-53 |
| ATGAGAACAGCGAGCAGA | AGG | Прямая | 71-88 | ||
| CTGTGTAATGCATGCTTG | TGG | Прямая | 112-129 | ||
| CGAAGCTTACTGAGGATT | TGG | Обратная | 194-177 | ||
| 2015 | Crispo [66] | GGCTGTGTAATGCATGCTTG | TGG | Прямая | 110-129 |
Примечание: зеленым и салатовым цветом выделены последовательности, совпадающие в разных экспериментах
 В ходе проведения анализа последовательности первого экзона гена миостатина с использованием специализированного электронного ресурса https://chopchop.cbu.uib.no было определено 32 возможных варианта целевой последовательности с прилегающим NGG мотивом (Рисунок 24).
В ходе проведения анализа последовательности первого экзона гена миостатина с использованием специализированного электронного ресурса https://chopchop.cbu.uib.no было определено 32 возможных варианта целевой последовательности с прилегающим NGG мотивом (Рисунок 24).
Рисунок 24 – Расположение предлагаемых целевых последовательностей в экзоне 1
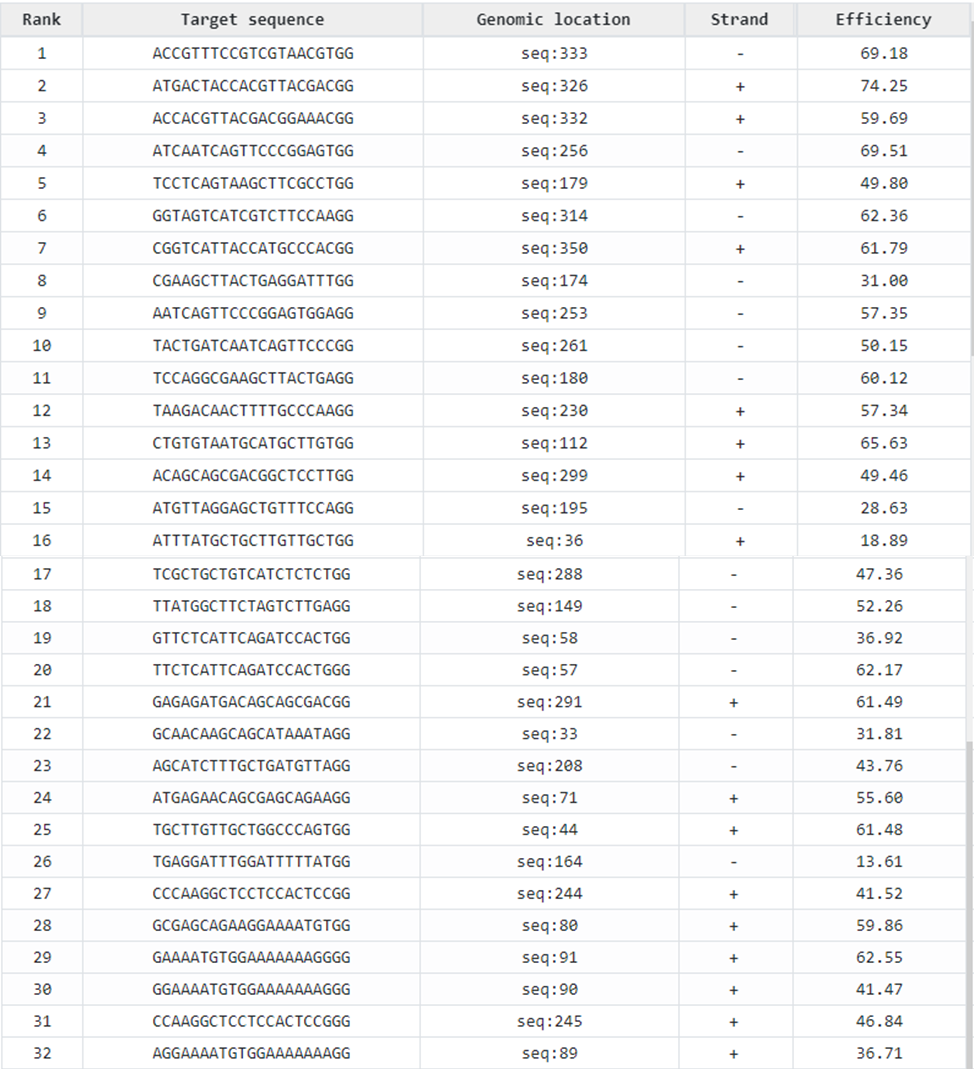
Инструменты используемого ресурса позволили получить информацию о нуклеотидной последовательности предлагаемых целевых сайтов, их локализации в гене, о том на какой из цепей (прямой или обратной) они расположены, а также позволили оценить их предполагаемую эффективность (Рисунок 25).

![]()
Рисунок 25 – Целевые последовательности, подобранные с использованием ресурса https://chopchop.cbu.uib.no
Две из подобранных нами целевых последовательностей (13 и 16, отмечены на рисунке) совпали с последовательностями из проанализированных ранее литературных источников и имели довольно удачное расположение с точки зрения возможного нарушения работы функциональных доменов гена. Они были отобраны для дальнейшей работы.
Используемый ресурс позволяет спрогнозировать возможные варианты репарации и редактирования цепи ДНК с предложенными целевыми последовательностями. Возможные варианты для целевой последовательности № 16 представлены на рисунке 26.
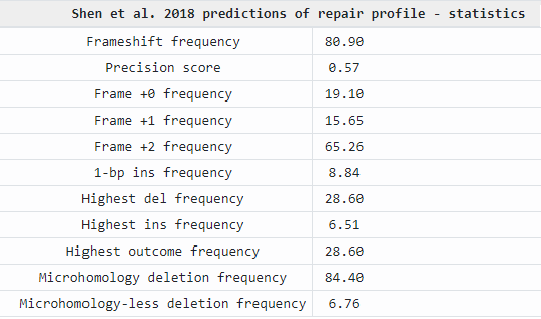
Рисунок 26 – Прогноз репарации при использовании целевой последовательности ATTTATGCTGCTTGTTGC
Возможные варианты для целевой последовательности № 13 представлены на рисунке 27.
 Рисунок 27 – Прогноз репарации при использовании целевой последовательности CTGTGTAATGCATGCTTG
Рисунок 27 – Прогноз репарации при использовании целевой последовательности CTGTGTAATGCATGCTTG
Таким образом, по результатам проведенного анализа для редактирования экзона 1 у овец определены две целевые последовательности (Таблица 10).
Таблица 10 – Целевые последовательности для редакции
| Наименование для внутреннего использования | Целевая последовательность (5’- 3’) | PAM | Цепь | Координаты целевой последовательности в гене |
| Guide1 | ATTTATGCTGCTTGTTGC | TGG | Прямая | 36-53 |
| Guide 2 | CTGTGTAATGCATGCTTG | TGG | Прямая | 112-129 |
6 Получение олигонуклеотидов и плазмидных векторов, кодирующих систему CRISPR/CAS
Для редактирования гена миостатина у овец использовался коммерческий набор GeneArt® CRISPR Nuclease Vector Kit, включающий в себя реактивы, плазмидные векторы и компетентные клетки.
Для использования GeneArt® CRISPR Nuclease Vector Kit были разработаны две пары одноцепочечных олигонуклеотидов с хвостами, комплементарными участкам линеаризованного вектора; один, два кодирующих целевую РНК CRISPR (целевая последовательность, прямая цепь) и два других – их комплементы (обратная цепь). Затем был проведен отжиг олигонуклеотидов прямой и обратной цепей для получения двухцепочечного олигонуклеотида (ds oligonucleotide), подходящего для клонирования в линеаризованный вектор, входящий в набор.
После выбора целевой последовательности из 18 пар оснований было проведено конструирование специфичных для crRNA олигонуклеотидных праймеров (Таблица 11).
Таблица 11 – Перечень синтезируемых последовательностей
| Наименование | Целевая последовательность (5’- 3’) | PAM | Цепь | Синтезируемая последовательности (5’- 3’) |
| Guide1 | ATTTATGCTGCTTGTTGC | TGG | Прямая | ATTTATGCTGCTTGTTGCGTTTT |
| Guide11 | gcaacaagcagcataaatcggtg | |||
| Guide 2 | CTGTGTAATGCATGCTTG | TGG | Прямая | CTGTGTAATGCATGCTTGGTTTT |
| Guide 22 | caagcatgcattacacagcggtg |
Для направленного клонирования ds-олигонуклеотида в нуклеазный вектор GeneArt® CRISPR к 3′-концам соответствующих одноцепочечных нуклеотидов было добавлено по 5 нуклеотидов. К 3′-концу олигонуклеотидов прямой цепи были добавлена последовательность GTTTT, комплементарная последовательности выступа, CAAAA, в линеаризованном нуклеазном векторе CRISPR и составляющая первые 5 оснований tracrRNA. Олигонуклеотид нижней цепи должен быть обратно комплементарен последовательности-мишени. К олигонуклеотидам обратной цепи, к 3′-концу были добавлены неуклеотиды CGGTG. Эта последовательность комплементарна последовательности выступа, CACCG, в линеаризованном нуклеазном векторе GeneArt® CRISPR и составляет последние 4 основания промотора U6 и первое основание, необходимое для стартового сайта транскрипции PolIII.
Далее был проведен отжиг двух одноцепочечных олигонуклеотидов для получения двухцепочечного олигонуклеотида с совместимыми концами для клонирования в нуклеазный вектор GeneArt® CRISPR.
Получение двухцепочечного олигонуклеотида
Необходимые материалы:
Forward strand oligonucleotide (200 µM in water or TE Buffer)
Reverse strand oligonucleotide (200 µM in water or TE Buffer)
50 µM stock of ds control oligonucleotide (thaw on ice)
10X Oligonucleotide Annealing Buffer
DNase/RNase-Free Water (supplied with kit)
1.5 mL sterile microcentrifuge tubes
95°C heat block
Процедура отжига
1. Вносили в чистую пустую пробирку при комнатной температуре следующие реагенты:
Forward strand oligonucleotide (200 µM) 5 µL
Reverse strand oligonucleotide (200 µM) 5 µL
10X Oligonucleotide Annealing Buffer 2 µL
DNase/RNase-Free Water 8 µL
Total volume 20 µL
2. Инкубировали пробирку при 95 ° С в течение 4 минут в термостате.
3. Доставали пробирку из термостата и давали реакционной смеси остыть до 25 ° C в течение 5–10 минут.
5. Центрифугировали пробирку в течение 5 секунд, осторожно перемешайте.
6. Для длительного хранения исходный раствор олигонуклеотида в концентрации 50 µM замораживали при -20 ° C.
7. После отжига одноцепочечных олигонуклеотидов и олигонуклеотидов, контролирующих клонирование, выполняли два 100‑кратных серийных разведения исходного раствора олигонуклеотидов с концентрацией 50 µM ds для приготовления исходного раствора олигонуклеотидов с концентрацией 500 nM ds (100-кратное разведение) и работали в дальнейшем с олигонуклеотидами ds в концентрации 5 nM. раствор (10 000-кратное разбавление).
Подготовка раствора dsОлигонуклеотидов 500 nM (stock solution)
Подготовили раствор двухцепочечных нуклеотидов в концентрации 500 nM путем разбавления стокового раствора нуклеотидов (концентрация 50 μM) в 100 раз.
1. Смешивали в микроцентрифужной пробирке:
50 μM ds oligonucleotide stock 1 μL
DNase/RNase-Free Water 99 μL
Общий обьем 100 μL
2. Тщательно перемешивали на вортексе. Раствор готов. Для длительного хранения 500 nM стоковый раствор двуцепочечных олигонуклеотидов замораживали при температуре –20°C.
Подготовка раствора ds Олигонуклеотидов 5 nM (working solution)
Подготовливали раствор двухцепочечных нуклеотидов в концентрации 5 nM путем разбавления стокового раствора нуклеотидов (концентрация 500 nM) в 100 раз.
1. Смешивали в микроцентрифужной пробирке:
500 nM ds oligonucleotide solution 1 μL
10X Oligonucleotide Annealing Buffer 10 μL
DNase/RNase-Free Water 89 μL
Общий обьем 100 μL
Оттаивание замороженных растворов олигонуклеотидов проводили на льду поскольку нагревание растворов двухцепочечных олигонуклеотидов приведет к их частичной денатурации и снижению эффективности клонирования.
Лигирование
После получения растворов двухцепочечных олигонуклеотидов в нужной концентрации клонировали синтезированные последовательности в нуклеазный вектор GeneArt® CRISPR. Вектор линеаризованный, имеет общую длину 9220 пар нуклеотидов, содержит последовательности, кодирующие белок Cas9 и зеленый флуоресцентный белок, сигналы ядерной локализации и ряд промоторов, обеспечивающих экспрессию CRISPR/Cas9 (Рисунок 28).
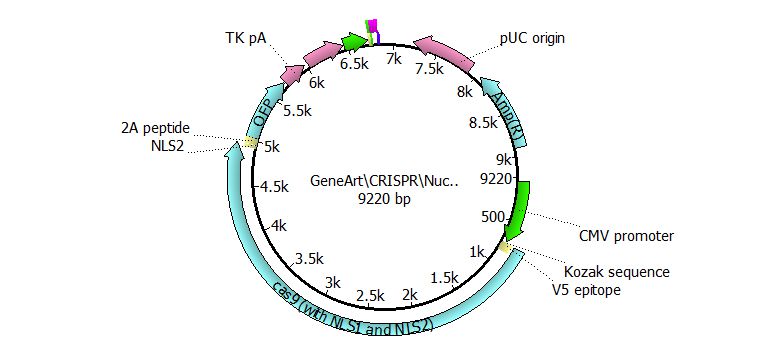
Рисунок 28 – Графическое представление нуклеазного вектора GeneArt® CRISPR
Вектор сконструирован таким образом, что синтезированный двуцепочечный олигонуклеотид благодаря специфичным последовательностям на 3’-концах встраивается в позицию crRNA между последовательностями, кодирующими U6 промотор и трансактивирующую РНК (Рисунок 29).

Рисунок 49 – Участок нуклеазного вектора GeneArt® CRISPR предназначенныей для встраивания последовательности crRNA
После клонирования в вектор двухцепочечного олигонуклеотида, полученного отжигом последовательностей Guide1 и Guide11и лигирования вектор замкнется в кольцо. Этот вид вектора получил название 111 (по используемым олигонуклеотидам) (Рисунок 30).
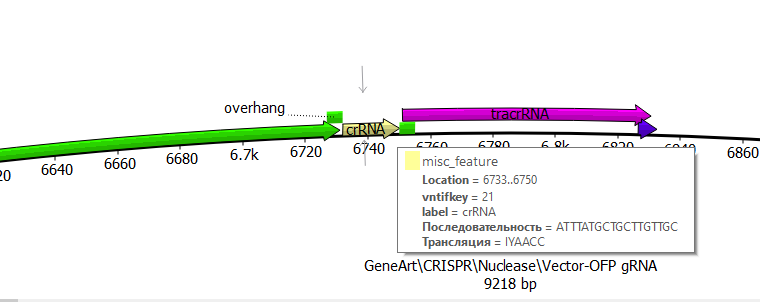
Рисунок 30 – Участок нуклеазного вектора GeneArt® CRISPR 111, содержащий специфичную направляющую последовательность
После клонирования в вектор двухцепочечного олигонуклеотида, полученного отжигом последовательностей Guide2 и Guide22 и лигирования вектор замкнется в кольцо.
Этот вид вектора получил название 222 (по используемым олигонуклеотидам) (Рисунок 31).

Рисунок 31 – Участок нуклеазного вектора GeneArt® CRISPR 222, содержащий специфичную направляющую последовательность
Необходимые материалы
Раствор двухцепочечных олигонуклеотидов (5 nM in 1X Oligonucleotide Annealing Buffer; оттаивание проводить на льду)
Раствор контрольных двухцепочечных олигонуклеотидов (5 nM in 1X Oligonucleotide Annealing Buffer; оттаивание проводить на льду)
Linearized GeneArt® CRISPR Nuclease Vector (thaw on ice before use)
5X Ligation Buffer (supplied with kit)
DNase/RNase-Free Water (supplied with kit)
T4 DNA Ligase (supplied with kit)
Контроль:
В качестве положительного контроля в эксперименте по лигированию использовался ds oligonucleotide поставляемый с набором.
Контрольный олигонуклеотид ds поставляется в виде исходного раствора 50 µM stock in 1X Oligonucleotide Annealing Buffer, и его необходимо повторно отжечь и разбавить в 10000 раз перед использованием в реакции лигирования.
Процедура лигирования
Общий объем смеси для лигирования каждого клонированного ds oligonucleoti составляет 20 μL.
Реакция лигирования проходит при комнатной температуре.
1. Внесили в чистую микроцентрифужную пробирку компоненты реакции в следующем порядке:
5X Ligation Buffer 4 µL
Linearized GeneArt ® CRISPR Nuclease Vector 2 µL
ds oligonucleotide (5 nM) 2 µL
DNase/RNase-Free Water 11 µL
T4 DNA Ligase 1 µL
Общий обьем реакции 20 µL
2. Тщательно перемешивали реакционную смесь пипетированием, не используя вортекс.
3. Инкубировали смесь в течение 10 минут при комнатной температуре (25–27 ° C).
Примечание: время инкубации может быть увеличено до 2 часов для увеличения выхода реакции.
4. Затем реакционную пробирку помещали на лед и переходили к трансформации компетентных клеток Transform One Shot ® TOP10 Competent E. Coli.
7 Клонирование генных конструкций в компетентных клетках.
Для наработки достаточного для экспериментов с эукариотическими клетками количества собранных нуклеазных векторов 111 и 222 выполнялось их клонирование в компетентных клетках E. Coli One ShotR, поставляемых в составе набора GeneArt® CRISPR Nuclease Vector Kit.
Для клонирования генных конструкций использовали следующие расходный материалы, стерильную посуду и оборудование:
1. Емкость со льдом
2. Чашки Петри (диаметром 5 см) одноразовые
3. Пробирки бактериологические (объемом от 10 до 20 мл)
4. Пипетки градуированные (пластиковые) (1, 2, 5, 10 мл)
5. Флаконы (пробирки) объемом 10 мл
6. Стерильный сосуд для слива
7. Шейкер-инкубатор
8. Термостат
9. Сосуд Дьюара
10. Твердотельный термостат
11. СО2 -инкубатор (термостат) с температурой 37О С
12. Центрифуга на 1000-2000 об /мин
13. Микроскоп инвертированный
14. Спектрофотометр
15. Ламинарный шкаф
16. Бактериальные петли
Также на данном этапе работы применяли растворы, питательные среды, сыворотки, антибиотики:
1. Компетентные клетки One ShotR
2. Плазмидная ДНК или продукты лигирования
3. Среда SOC
4. LB агар
5. LB бульон
6. Ампициллин
7. Стерильный глицерин
Трансформацию клеток выполняли по следующей схеме:
1. Поместили на лед пробирки с компетентными клетками до полного размораживания содержимого из расчета одна пробирка на трансформацию (50 мкл клеток One ShotR). Примерно 10 – 15 минут.
2. Добавили в каждую пробирку 3 мкл образца ДНК (плазмидная ДНК, продукты лигирования). Аккуратно перемешали содержимое легким встряхиванием.
3. Немедленно перенесли пробирки на лед. Инкубировали пробирки на льду в течение 30 минут.
4. Затем перенесли пробирки в твердотельный термостат (42 °С) и инкубировали в течение 30 секунд. Без встряхиваний.
5. Из твердотельного термостата пробирки (БЫСТРО!) перенесли на лед и инкубировали 3 минуты.
6. Добавили 250 мкл среды SOC (возможно использование LB бульона) комнатной температуры, перемешали содержимое.
7. Поместили пробирки в шейкер-инкубатор и инкубировали в течение 1 часа при 37 °С и 225 об/мин.
8. Высеяли 50 мкл реакционной смеси на предварительно нагретые чашки Петри с LB-агаром, содержащие 100 мкг/мл ампицилина. Оставшуюся часть реакции нанесли на вторую чашку Петри с LB-агаром (предварительно подогретую). Чашку Петри инкубировали в течение ночи при 37 °С. Поскольку вектор Linearized GeneArt ® CRISPR Nuclease Vector несет ген устойчивости к ампицилину, бактерии, успешно пережившие трансформацию и принявшие плазмиды 111 и 222, выжили и в дальнейшем образовали колонии. Остальные клетки погибли.
9. На следующие сутки отбирали 2-3 колонии (рисунок 31), помещали их в бульон и ставили в термостат на доращивание (14-18 часов). После чего выделяли плазмидную ДНК – чистый вектор для трансфекции эукариотических клеток. Перед выделение ДНК готовили библиотеку клеток (по протоколу криоконсервация библиотеки клеток).
10. Перед выделением ДНК проверяли бульон на мутность на спектрофотометре при длине волны 600 нм («мутность суспензии»).
Рисунок 32 – Колонии бактерий в чашке Петри.
Криоконсервацию библиотеки клеток осуществляли по следующему протоколу:
В ламинарном шкафу бактериальной петлей набрали колонию бактерий и перенесли ее в пробирку с 2 мл LB бульона. Инкубировали в течение ночи (14–18 ч) при 37 °С в шейкер инкубаторе 250 об. мин. Пока культура не достигнет стационарной фазы (среда станет мутной). Смешали 0,85 мл культуры с 0,15 мл стерильного глицерина и перенесли в криопробирку. Перемешали в шейкер-инкубаторе. Концентрация глицерина может варьировать от 30 до 50 %. Далее пробирки перенесли в сосуд Дьюара и заморозили при -80 °С.
8 Выделение и очистка плазмидных векторов, секвенирование встроенных участков с кодом гайдовой РНК, оценка качества выделенной ДНК и полученных конструкций.
Для выделения плазмидной ДНК из культуры клеток E. Coli применяли набор Plasmid Midiprep 2.0 производитель компания «Евроген». Выделение ДНК основано на применении микроцентрифужных колонок Midi с сорбционными стекловолокнистыми мембранами, предназначенных для центрифугирования. Основные свойства набора: емкость колонки составляет 500 мкг плазмидной ДНК; спектральный показатель препарата плазмидной ДНК A260/А280> 1,85; время выделения от 1 часа 30 мин.
Перед выделением ДНК бактериальную культуру необходимо выращивать примерно 16–20 часов в термостатируемом шейкере при 37°C, при постоянном перемешивании 200–250 об/мин. Следует избегать “перероста” бактериальной культуры, так как это может привести к большому количеству погибших клеток и к разрушению плазмид или загрязнению их хромосомными ДНК. плотность бактериальной культуры на спектрофотометре (OD600). Плотность бактериальной культуры контролировали с использованием спектрофотометра (OD600). По окончании роста на длине волны 600 нм показатель OD600 должен быть не менее 1,4‑1,6, плотность клеток 3–5 x 109 клеток/мл. Для плазмид с разной копийностью для эффективного выделения используется различный объем суспензионной культуры (Таблица 12).
Таблица 12 – Выход ДНК для плазмид.
| Копийность плазмиды | Объем среды | Предполагаемый выход ДНК |
| высокая | 25–50 мл | 300–500 мкг |
| низкая | 50–150 мл | 100–350 мкг |
Для выделения плазмидной ДНК использовали реактивы из набора Plasmid Midiprep 2.0: РНКаза A (лиофилизированная), ресуспендирующий раствор, лизирующий раствор, нейтрализующий раствор, промывочный раствор (концентрат), элюирующий раствор – 5 mM Tris-HCl 7,5-pH, раствор для удаления эндотоксинов.
Также использовали следующие материалы и оборудование:
- Пробирки объемом 50 мл для центрифугирования;
- Микроцентрифужные пробирки на 2 мл, Eppendorf;
- Штатив для пробирок на 50 мл;
- Охлаждаемая центрифуга с ротором для пробирок на 50 мл, относительное центробежное ускорение не менее 3 000 g;
- Настольный твердотельный термостат;
- Этиловый спирт (96%).
Приготовление растворов для выделения плазмидной ДНК: во флакон с концентрированным промывочным раствором добавили 210 мл 96% этилового спирта, перемешали. Чтобы растворить «РНКазы A» добавили в пробирку 0,5 мл ресуспендирующего раствора. После растворения перенесли полученный раствор во флакон с ресуспендирующим раствором», перемешали.
Выделение ДНК осуществляли согласно протоколу «Plasmid Midiprep 2.0». Перед началом работы включили охлаждение в центрифуге; охладили нейтрализующий раствор при +4°C; для улучшения элюции нагрели флакон с элюирующим раствором до +50°C. Далее перенесли ночную бактериальную культуру в пробирку на 50 мл, осадили клетки центрифугированием (3 773 g) при температуре 4°C в течение 15 минут. Полностью удалили осветленную культуральную среду. Центрифугировали пробирку с осадком 30 секунд. Пипеткой удалили остатки культуральной среды. Добавили 4 мл ресуспендирующего раствора с «РНКазой А» к осадку и тщательно ресуспендировали на вортексе до гомогенности суспензии. Добавили 4 мл лизирующего раствора, перемешали содержимое пробирки 6‑кратным переворачиванием, пока лизат не стал вязким и полупрозрачным. Инкубировали при комнатной температуре не более 5 минут, больше 5 минут не рекомендуется, иначе плазмидная ДНК денатурирует. Добавили 4 мл предварительно охлажденного нейтрализующего раствора, перемешали содержимое, переворачивая пробирку до образования равномерной творожистой взвеси. Центрифугировали пробирку 30 минут для осветления лизата (3 773 g) при комнатной температуре. Поместили спин-колонку в пробирку на 50 мл, пробирку установили в штатив для более удобной работы. Добавили в центр спин-колонки 0,5 мл раствора для удаления эндотоксинов, чтобы уравновесить колонки. Перенесли осветленный бактериальный лизат в колонку. Центрифугировали пробирку со спин-колонкой 1 минуту не более 1850 g при комнатной температуре. В процессе центрифугирования плазмидная ДНК собирается на фильтре колонки. Далее удалите фильтрат из пробирки. В колонку добавили 2 мл раствора для удаления эндотоксинов. Центрифугировали пробирку с колонкой 1 минуту при 1 850 g при комнатной температуре. Добавили 15 мл промывочного раствора со спиртом в колонку. Центрифугировали пробирку с колонкой 1 минуту при 3 773 g при комнатной температуре. Удалили фильтрат из пробирки. Еще раз повторили процедуру: добавили 15 мл промывочного раствора со спиртом в колонку. Центрифугировали пробирку с колонкой 1 минуту при 3773g при комнатной температуре и снова удалили фильтрат из пробирки. Центрифугировали пробирку с колонкой 10 минут при 3773 g. Поместили спин-колонку в новую пробирку на 50 мл. Чтобы остатки спирта на фильтре не снизили выход ДНК и не повлияли на спектрофотометрические измерения спин-колонки посушили при комнатной температуре 8 минут. Нанесли на фильтр спин-колонки 2–3 мл предварительно нагретого до 50°C элюирующего раствора. Инкубировали 2 минуты при комнатной температуре. Центрифугировали колонку 1 минуту для сбора очищенной ДНК при 3 773 g, элюат собирается в пробирки.
Концентрацию плазмидной ДНК проверяли на приборе Nanophotometer «ImpLEN» (Рисунок 32). Встроенный вортекс обеспечивает однородность образца с наивысшей степенью точности с наименьшим объемом образца всего 0,3 мкл. Линзу прибора аккуратно очистили салфеткой, чтобы избежать ложноположительного или ложноотрицательного результата. В качестве бланка использовали элюирующий раствор (1,5 мкл), в котором была элюирована полученная плазмидная ДНК. На линзу нанофотометра нанесли 1,5 мкл плазмидной ДНК. Согласно инструкции, соотношение А260/280 должно быть равным 1,7-1,9… ng/ul. Соотношение А260/230 имеет второстепенную важность, но должно вписываться в пределы 1,5-3,0 ng/ul (Таблица 13).
Таблица 13 – Проверка качества выделенной плазмидной ДНК.
| ДНК | Конц. | Ед. | А230 | А260 | А280 | А320 | А260/
А280 |
А260/
А230 |
| Blank (эл. р-р ) | 0,0000 | ng/ul | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 |
| Crispr Ovis 111 | 31,650 | ng/ul | 0,351 | 1,086 | 0,557 | 0,013 | 1,972 | 3,175 |
| Crispr Ovis 111. | 31,600 | ng/ul | 0,319 | 1,077 | 0,544 | 0,005 | 1,989 | 3,414 |
| Crispr Ovis 222 | 33,800 | ng/ul | 0,370 | 1,067 | 0,535 | 0,011 | 2,015 | 2,942 |
| Crispr Ovis 222. | 33,900 | ng/ul | 0,334 | 1,080 | 0,563 | 0,002 | 1,922 | 3,247 |
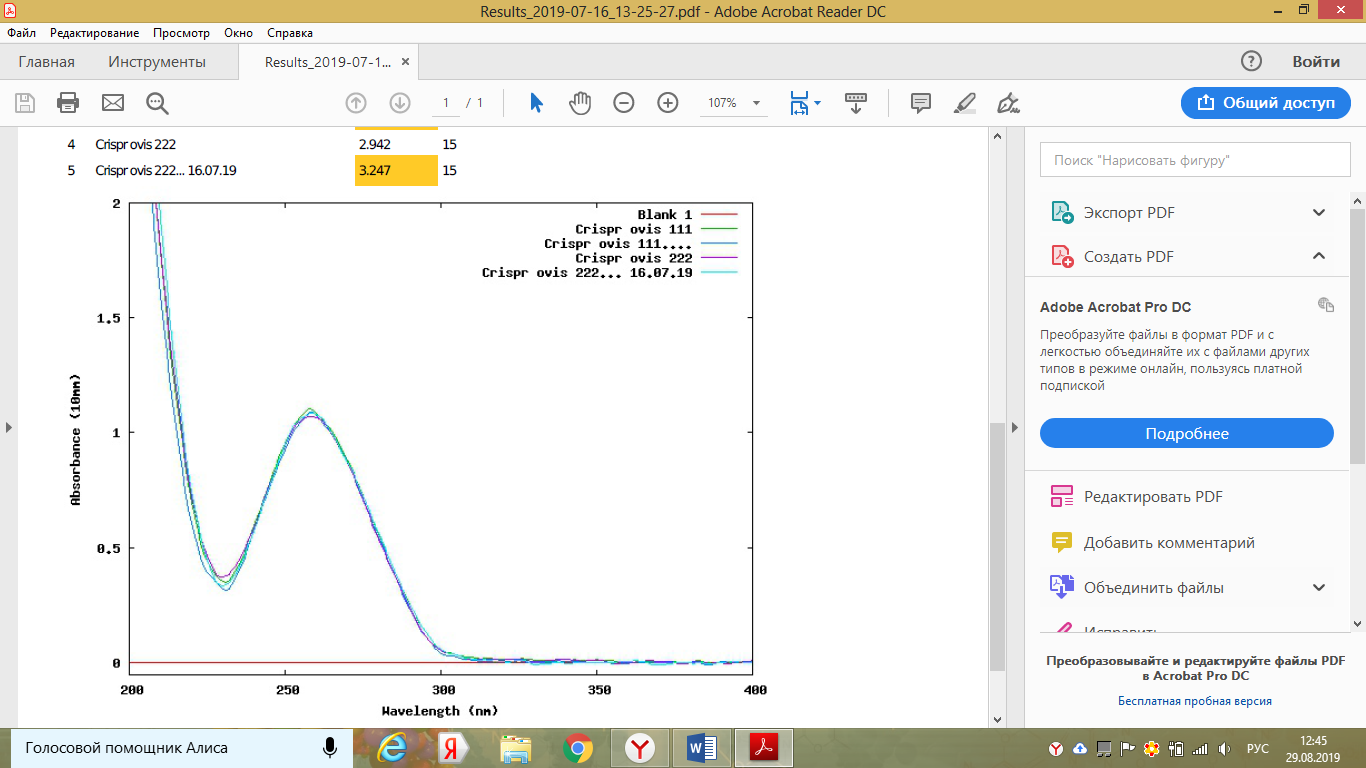 По данным таблицы видно, что качество выделенной плазмидной ДНК, хорошее, согласно инструкции, совпадает практически со всеми критериями.
По данным таблицы видно, что качество выделенной плазмидной ДНК, хорошее, согласно инструкции, совпадает практически со всеми критериями.
Рисунок 33 – Концентрация плазмидной ДНК.
Однако, соотношение А260/А230 превышает в трех пробах. Поэтому, для дальнейшей трансфекции с плазмидными векторами, необходимо провести очистку образцов от примесей и увеличить концентрацию плазмидной ДНК.
Остаточные эндотоксины удалили путем переосаждения препарата ДНК. Не использовали изопропанол для переосаждения, т.к. следы изопропанола очень сильно снижают эффективность трансфекции. Центрифугирование при переосаждении на всех этапах проводили при комнатной температуре на скорости 12 000 об/мин. Рекомендации по выбору режима центрифугирования для очистки плазмидной ДНК таковы, эффективность центрифугирования образца определяется относительным ускорением центрифуги. Оно зависит от частоты вращения (RPM, rotation perminute) и радиуса центрифугирования. Относительное ускорение центрифуги (RCF) задается как кратное от ускорения свободного падения (g) и характеризует условия центрифугирования на любой центрифуге, вне зависимости от радиуса ротора. Величины RCF и RPM связаны между собой следующей формулой:
??? = 1,118 × 10-5 × n2 × ?,
где ??? – относительное ускорение, ? – радиус ротора в см, n – частота вращения в оборотах в минуту (???).
Роторы различных моделей настольных центрифуг обладают разным радиусом, поэтому одинаковое количество оборотов в минуту для разных центрифуг будет приводить к разному относительному ускорению, то есть разной эффективности центрифугирования. Для переосаждения плазмидной ДНК использовали пробирки Eppendorf и высокоскоростную центрифугу «СМ-50» производитель компания ELMI (Латвия). Центрифуга СМ-50 является одной из самых компактных и мощных мини-центрифуг.
Переосаждение проводили согласно протоколу. Отобрали аликвоту по 0,5 мл очищенной на спин-колонке плазмидной ДНК по двум участкам гена миостатина в микроцентрифужные пробирки объемом 2 мл (10 штук – по 5 пробирок на последовательность). Добавили 50 мкл 3M ацетата натрия pH 5,2 и 1,4 мл 96% этанола, перемешали на вортексе. Инкубировали 2 минуты при комнатной температуре. Центрифугировали образцы в настольной центрифуге 10 минут. Аккуратно удалили осветленную жидкость, чтобы не захватить на дне пробирки беловатый осадок. Аккуратно добавили 0,5 мл 70% этилового спирта. Поместили пробирку в ротор в том же положении. Центрифугировали образец в настольной центрифуге 10 минут. Аккуратно удалили спиртовую фазу и подсушили осадок при комнатной температуре 8 минут. Осадок стал полностью прозрачным (бесцветным). Растворили осадок в 50 мкл элюирующего раствора. Для проверки качества переосажденной ДНК использовали также прибор Nanophotometer «ImpLEN» (Рисунок 34).
Концентрация плазмидной ДНК увеличилась в 10-20 раз, что позволяет использовать аликвоту для постановки трансфекции (Таблица 14). По данным таблицы видно, что соотношение А260/А280 и А260/А230 вписываются в пределы нормы.
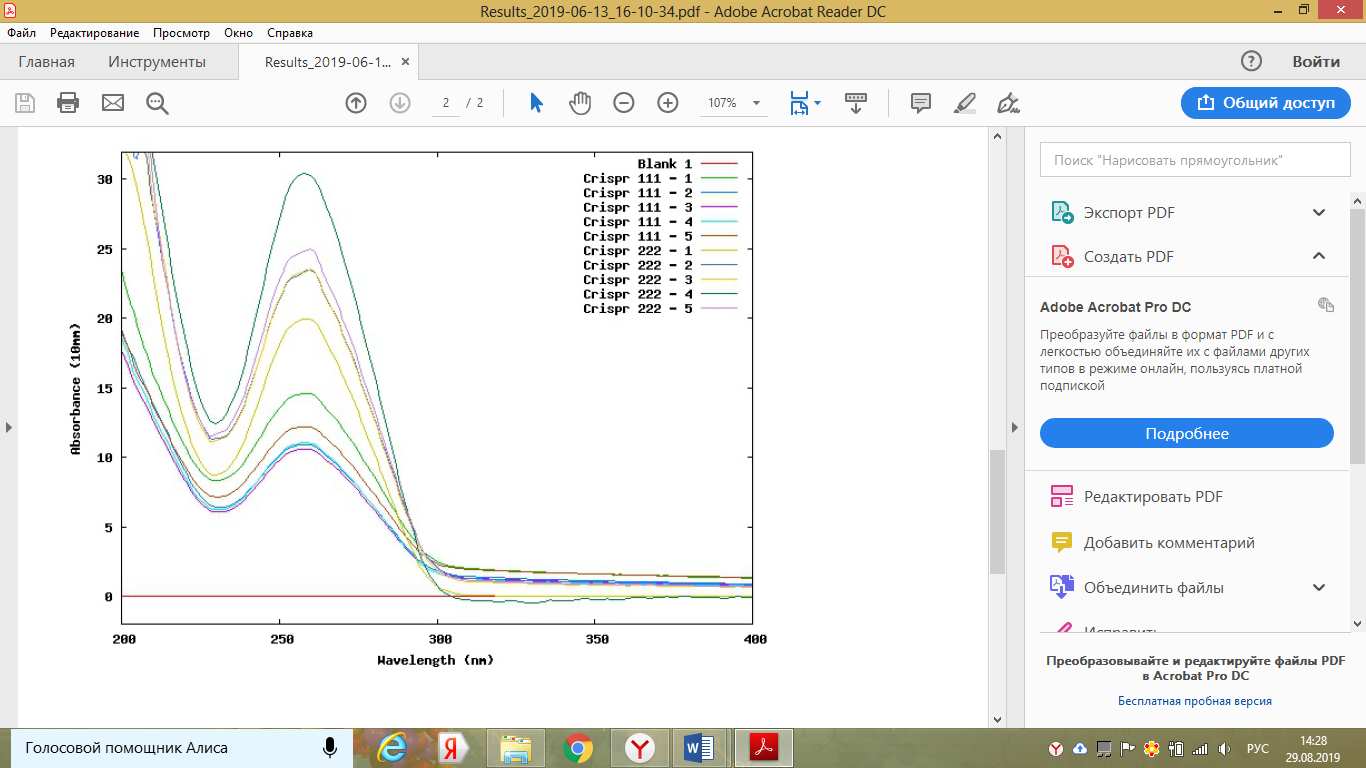 Рисунок 34 – Концентрация очищенной плазмидной ДНК.
Рисунок 34 – Концентрация очищенной плазмидной ДНК.
Таблица 14 – Оценка качества очищенной плазмидной ДНК.
| ДНК | Конц. | Ед. | А230 | А260 | А280 | А320 | А260/
А280 |
А260/
А230 |
| Blank (эл. р-р ) | 0,0000 | ng/ul | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 |
| Crispr Ovis 111 | 633,70 | ng/ul | 8,342 | 14,55 | 8,635 | 1,876 | 1,875 | 1,960 |
| Crispr Ovis 111. | 476,95 | ng/ul | 6,424 | 10,87 | 6,436 | 1,331 | 1,869 | 1,873 |
| Crispr Ovis 111 | 467,90 | ng/ul | 6,059 | 10,57 | 6,232 | 1,212 | 1,864 | 1,931 |
| Crispr Ovis 111. | 492,35 | ng/ul | 6,241 | 11,01 | 6,430 | 1,163 | 1,870 | 1,939 |
| Crispr Ovis 111 | 518,00 | ng/ul | 7,157 | 12,19 | 7,348 | 1,830 | 1,877 | 1,945 |
| Crispr Ovis 222 | 994,10 | ng/ul | 8,724 | 19,92 | 10,33 | 1,038 | 1,932 | 2,289 |
| Crispr Ovis 222. | 1000,6 | ng/ul | 11,35 | 21,49 | 11,83 | 1,179 | 1,915 | 2,194 |
| Crispr Ovis 222 | 998,40 | ng/ul | 11,20 | 21,52 | 11,71 | 1,385 | 1,922 | 2,206 |
| Crispr Ovis 222. | 997,70 | ng/ul | 11,45 | 20,24 | 11,01 | 1,389 | 1,868 | 2,386 |
| Crispr Ovis 222 | 1000,3 | ng/ul | 11,66 | 24,96 | 13,42 | 1,057 | 1,933 | 2,254 |
Для того чтобы оценить качество полученных конструкций и убедиться, что направляющие нуклеотидные последовательность встроились в вектор в правильной ориентации и без каких-либо ошибок было проведено секвенирование плазмидных векторов выделенных из колоний клеток трансформантов.
Секвенирование нуклеазной конструкции CRISPR выполнялось с использованием прямого праймера U6, входящего в комплект GeneArt® CRISPR Nuclease Vector Kit — 5′-GGACTATCATATGCTTACCG -3′. Последовательности, полученные в ходе секвенирования векторов, выделенных из клеток трансформантов представлен в Таблице 15.
Таблица 15 – Фрагменты последовательности векторов 111 и 222, выделенных из различных колоний клеток-трансформантов
| Колония | Вектор |
| 111 | |
| А111 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGATTTATGCTGCTTGTTGCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| В111 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGATTTATGCTGC- — GTTGCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| 222 | |
| А222 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGCTGTGTAATGCATGCTTGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| В222 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGCTGTGTAATGCATGCTTGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
Из таблицы видно, что в колониях A222 и В222, трансформированных с использованием вектора 222 последовательность вектора соответствует ожидаемой и не различается, направляющая последовательность CRISPR (в таблице выделена жирным шрифтом) имеет правильную ориентацию. В колонии А111, трансформированной с использованием вектора 111 последовательность вектора также не нарушена и имеет правильную ориентацию. Однако вектор, выделенный из колонии В111, трансформированной с использованием вектора 111, содержит делецию TT в области направляющей последовательности и таким, образом не может быть использован в дальнейшей работе.
Таким образом, для наращивания количества работоспособного вектор 111 в дальнейшей работе использовалась колония A111, для наращивания количества работоспособного вектор 222 использовались колонии А222 и В222.
9 Получение культур фибробластов, полученных от экспериментальной группы овец.
В соответствии с нашими предварительными экспериментами получение значительного количества фибробластов (~108 клеток) из небольшого (3–4 мм3) биопсийного образца делится на два этапа. Первый этап заключается в получении первичной культуры фибробластов и существенно зависит от состава культуральной среды, в которой должны содержаться ростовые факторы, обеспечивающие размножение эксплантированных в низкой плотности клеток. Второй этап, заключающийся в экспансии первичной культуры в условиях высокой плотности посева (~104 кл./см2), также определяется ростовыми компонентами культуральной среды, но в значительно меньшей степени, по‑видимому, в силу способности самих фибробластов синтезировать ряд факторов роста: фактор роста кератиноцитов (keratinocyte growth factor, KGF), сосудистый эндотелиальный фактор роста (vascular endothelial growth factor, VEGF), инсулиноподобный фактор роста (insulin-like growth factor, IGF) и фактор роста гепатоцитов (hepatocyte growth factor, HGF). Традиционно источником ростовых факторов в культуральной среде является фетальная сыворотка крупного рогатого скота (fetal bovine serum, FBS). Свойства сыворотки в силу множества факторов, влияющих на ее состав при производстве, могут значительно варьировать, что отражается на получении первичной культуры фибробластов [78–80].
Нашей задачей явилось получение однослойных первичных культур клеток фибробластов овцы методом трипсинизации. Исследования проводили по следующей схеме:
1. Биопсия кожи. Фибробласты можно получить из многих тканей и органов. Проще всего – из дермального слоя кожи. Удобнее проводить биопсию кожи у основания ушной раковины животного. Для этого кожу дезинфицировали 70 % этиловым спиртом. После дезинфекции кожу захватывали гемостатическим зажимом и скальпелем срезали кусочек кожи над браншами зажима. При правильно проведенной биопсии, захватывающей эпидермис и сосочковый слой кожи, кровотечение минимальное. Отсеченный фрагмент кожи (5х5мм) погружали во флакон с предварительно подогретым до Т=37 °С фосфатным буфером (PBS) и антибиотиком. Плавными движениями перемешать содержимое флакона. Образец хранить до дальнейших манипуляций при Т=37 °С не более 30 минут. Далее работу вели в ламинарном боксе. Работы в ламинарном боксе проводили следующем образом: Внутреннюю поверхность ламинарного бокса, пробирки с питательными средами, пипетки, пробирки протирали 70%-м спиртом. После включения ультрафиолета включили вытяжку, проветрили помещения в течение 15-20 минут. Для надежности стерилизации перед началом работы помещение лаборатории и внутреннее пространство ламинара облучают УФ‑лучами. Облучение ультрафиолетовыми лучами (260 нанометров) — наиболее часто используется в лабораториях для стерилизации помещений, настольных боксов. При длительном воздействии эти лучи вызывают гибель всех бактерий. Бактерии погибают очень быстро, а споры грибов значительно медленнее. Поэтому в боксах устанавливают бактерицидные лампы БУФ-15 или БУФ-30, которые включаются на 30 минут за 1 час до работы. Кроме того, проводили профилактическое облучение боксов в течение 2 часов.
В ламинарный бокс поместили сосуды с культурами клеток. Руки тщательно протирали 70%-м спиртом. При работе со стерильной посудой и материалом необходимо помнить, что нельзя проносить руки над открытой стерильной поверхностью. Все инструменты перед использованием и в процессе работы стерилизуют, для чего помещают в сосуд с 96%-м спиртом и обжигают внесением в пламя спиртовки до полного выгорания спирта. В целях избегания воспламенения спирта в сосуде не разрешается вносить инструменты после обжигания в пламени спиртовки и вновь в сосуд со спиртом без их остывания в течение 10-15 секунд. В случае воспламенения сосуда со спиртом необходимо сразу накрыть сосуд плотным полотенцем и прижать его на несколько секунд, перекрывая доступ воздуху к горящей поверхности. По окончании работы колбы, пробирки и чашки Петри подписывали и помещали в условия культивирования. Поверхность ламинарного бокса протирали 70%-м спиртом, инструменты и материалы упаковывали и помещали на стерилизацию. При работе с культурами клеток в ламинарном боксе необходимо соблюдать техника безопасности. При выполнении исследований по изучению: способов приготовления питательных сред; подготовки посуды микроорганизмов; для культивирования изучению состава питательных сред и техники посевов культур микробов в жидкие, полужидкие и на плотные среды необходимо неукоснительно соблюдать правила, обязательные при работе с культурами микроорганизмов, газовыми горелками, сосудами, работающими под давлением (автоклавы), лабораторной стеклянной посудой, электрооборудованием.
2. Механическая и ферментативная дезагрегация ткани. Полученный биоптат кожи измельчали на кусочки размером 1–3 мм, отмывали от эритроцитов раствором PBS с антибиотиками. Для дезагрегации ткани использовали 0,25 % трипсин. В нем выдерживали кусочки ткани в термостате при температуре 37 °С в течение 30 мин. В результате получали густую суспензию разволокненной ткани.
3. Получение первичной культуры фибробластов кожи. Полученную суспензию клеток центрифугировали на скорости 2000 об. в течение 5 минут.
Необходимое оборудование и расходный материал:
1. Культуральные флаконы (матрасы) для культивирования клеток.
2. Чашки Петри (диаметром 5 см) одноразовые
3. Пробирки бактериологические (объемом от 10 до 20 мл)
4. Пипетки градуированные (пластиковые) (1, 2, 5, 10 мл)
5. Флаконы (пробирки) объемом 10 мл
6. Стерильный сосуд для слива
7. Пинцеты анатомические
8. Ножницы медицинские (глазные) с прямыми и закругленными браншами
9. Игла-пробойник
10. Камера Горяева с покровными стеклами
11. СО2 -инкубатор (термостат) с температурой 37О С
12. Центрифуга на 1000-2000 об /мин
13. Микроскоп инвертированный
В исследовании использовали растворы, питательные среды, сыворотки, антибиотики, такие как:
— фосфатный буфер (PBS), физ. раствор
— раствор Версена с 0,25% раствором трипсина (1 части трипсина + 3 части версена).
— дезинфицирующие растворы: 70% этиловый спирт
— ростовые среды – среда ДМЕМ
— сыворотка крови плодов крупного рогатого скота (ЭТС). Перед первым использованием разморозить и прогреть при +56ОС — 30 мин или при +45ОС – в течение 1 часа. Рабочее количество хранится при +4ОС, остальное количество при –20ОС.
— глютамин
— антибиотики – пенициллин и стрептомицин 100 мкг/мл.
Полная среда (с сывороткой) готовится перед ее применением и не более чем на 1 месяц работы. Для большинства перевиваемых линий клеток количество сыворотки составляет 5-10 % объема среды роста.
Величина рН ростовых сред должна быть в пределах 7,5-7,6. Все жидкие компоненты, используемые при культивировании, должны быть прозрачными и не иметь осадка.
Среду для культивирования фибробластов готовили из стерильной среды ДМЕМ с добавлением 10% фетальной телячьей сыворотки, пенициллин и стрептомицин 100 мкг/мл. В среду также вносят глютамин 0,3 мг/мл.
Биоптат после взятия отмывали фосфатным буфером. Переместили в пробирку эппендорф, центрифугировали. Удалили надосадочную жидкость. Далее осадок ресуспендировали в 1 мл полной питательной среды, в которую входят компоненты такие как: среда ДМЕМ, сыворотка, антибиотик. Повторяли процедуру 3 раза до полного отмывания. Порядок подготовки фибробластов для культивирования представлен на рисунке 35.
 Рисунок 35 – Подготовка фибробластов для культивирования
Рисунок 35 – Подготовка фибробластов для культивирования
Затем супернатант перенесли на дно предварительно подогретую, стерильную чашки Петри, при этом равномерно распределив его по всей поверхности. В чашке Петри заранее наливали примерно 4 мл культуральной среды и поставили в термостат на инкубацию при 37 °С в атмосфере, содержащей 5 % СО2 (рисунок 36).
Каждые 48 часов (2-е суток) чашки Петри просматривали с помощью инвертированного микроскопа и для оценки качества культуры делали серию снимков. Отслеживали рост клеток, а также следили затем, чтобы в культуре не произошла контаминация, так как одной из основных причин замедления роста и гибели клеток является появление в культуре бактерий или грибов.
Рисунок 36 – Культуральная среда с супернатантом.
Культуру клеток фибробластов инкубировали в СО2–инкубаторе Binder С–150 (Binder, Германия) в течение 8 дней. В ходе нашего исследования становится видно, что на 5 сутки культивирования дне сосуда появляются единичные фибробласты – плоские клетки веретеновидной формы с отростками, прозрачной цитоплазмой и плоским овальным ядром представлены на рисунке 37.

 Рисунок 37 – Культура клеток фибробластов на 5-е сутки
Рисунок 37 – Культура клеток фибробластов на 5-е сутки
Рисунок 38 – Культура клеток фибробластов на 8-е сутки
На 8 сутки культивирования на дне чашки Петри количество прикрепленных клеток увеличилось (Рисунок 39). Для того, чтобы процесс роста клеток был активный необходима еженедельная смена среды в чашке Петри.
Смену среды осуществляли в ламинарном боксе по протоколу:
- Перед работой обрабатывали руки спиртом. Далее из чашки Петри отбирали 1 мл старой среды в эппендорф пробирку, центрифугировали 3 минуты.
- Среду из чашки Петри сливали в чашку для отходов.
- В чашку Петри наливали PBS (с антибиотиком) и аккуратно промывали, сливали в пустую чашку Петри для отходов.
- В чашку Петри заливали культуральную среду, промывали и сливали в чашку Петри для отходов.
- Затем в чашку Петри добавляли новой культуральной среды с антибиотиком и сывороткой. Также добавляли 0,5 мл отцентрифугированной старой среды. Далее среду с фибробластами культивировали в термостате. На рисунке 38 представлена культура клеток на 13-е сутки.

Рисунок 39 – Культура клеток на 13-е сутки
Для получения линии фибробластов, монослой пересевают, используя трипсинизацию (трипсин 0,05%). Трипсинизированные и перенесенные в новые сосуды клетки становятся вторичной культурой, и по формальным признакам такая культура носит название линии клеток.
Под микроскопом просмотрели чашку Петри с культурой клеток. Культуру клеток с конфлюентностью свыше 60% готовим к пересеву (Рисунок 40). Работу по пересеву культуры вели в ламинарном боксе. Ламинарный бокс предварительно облучили бактерицидными ультрафиолетовыми лампами (20-30 минут), весь необходимый расходный материал обработали 70% спиртом. Для предотвращения загрязнения культур не следует использовать одни и те же бутыли со средой и реагентами при работе с различными культурами в чашках Петри. Пипетки, имевшие контакт с флаконами и бутылями, не следует повторно использовать для отбора среды из соответствующих емкостей.
 Рисунок 40 – Культура клеток на 15-е сутки
Рисунок 40 – Культура клеток на 15-е сутки
Расположение элементов в ламинарном боксе для культуры клеток обычно придерживается следующих правил, которые могут быть изменены, чтобы включить дополнительные элементы, используемые в конкретных приложениях. Широкое, чистое свободное пространство с рабочими принадлежностями для культуры клеток в центре. Дозаторов располагается спереди в правой части, где его можно легко достать. Реагенты и среды в задней части справа, что позволяет легко перемешивать. Подставка для пробирок для дополнительных реагентов располагается посередине сзади.
Пересев клеток чаще всего выполняется на 14-е – 16-е сутки по следующему протоколу:
Из чашки Петри отбирали 1 мл «старой» среды в пробирку эппендорфа. Центрифугировали 5 минут при 300 G, затем отбирали 0,5 мл отцентрифугированной среды и оставляли ее, для последующего переноса ее в культуральный матрас.
- Оставшуюся старую среду из чашки Петри утилизировали (сливали) в чашку для отходов. В чашку Петри наливали фосфатный буфер (PBS) с антибиотиком, хорошо промывали. После промывки буфер сливали в пустую чашку Петри для отходов.
- В чашку Петри с культурой клеток заливали раствор версен + трипсин (2,5 мл) и ставили на 5 минут в термостат (или на термостолик) при температуре 37°С. Наблюдали под микроскопом за снятием монослоя каждые 7 минут. Процесс длится 15 – 30 минут.
- Из чашки Петри раствор с клетками переносили в эппендорф пробирку.
- Центрифугировали пробирку на скорости 2000 оборотов в течение 3 минут. Удалили надосадочную жидкость.
- Внесли в пробирку с клетками 600 мкл среды DMEM. Содержимое пробирки плавно перемешали.
- Центрифугировали пробирку на скорости 2000 оборотов в течение 3 минут. Удалили надосадочную жидкость.
- Внесли в пробирку с клетками 600 мкл среды DMEM для культивирования. Содержимое пробирки плавно перемешали.
- Содержимое из пробирки эппендорф переносили в чашку Петри для культивирования.
- Затем в чашку Петри добавляли 0,5 мл старой среды (пункт 2.) Заранее в чашку Петри (с желатином) налили 3 мл новой среды и поставили в термостат при температуре 37 °С. Для наращивания культуральной массы можно использовать культуральные флаконы.
Ежедневно проводили просмотр культуральных сосудов для оценки цвета среды и ее прозрачности. Резкое изменение цвета и появление мути может свидетельствовать о бактериальном или грибковом заражении. Если цвет индикатора среды не изменяется после пересева это служит показателем благоприятных условий среды для размножения клеток фибробластов. Если индикатор сдвигается в сторону светло-малинового, это может быть связано с отсутствием размножения клеток, с недостаточным количеством для данного объема или гибелью, а также контаминацией клеточной культуры. На вторые сутки при помощи инвертированного микроскопа оценивали морфологию клеток и слоя в целом. Микроскоп инвертированный — это оптический микроскоп, отличающейся тем, что объектив располагается под наблюдаемым предметом, а конденсор — сверху. Направление хода лучей, прошедших сверху вниз через объектив, изменяется системой зеркал, и в глаз наблюдателя они попадают, как обычно, снизу-вверх. В ходе работы при оценке появилось большое количество плавающих клеток округлой формы, способных к делению. Единичные клетки начали прикрепляться к подложке (Рисунок 41).

Рисунок 41 – Культура клеток на 2-е сутки после пересева
При благоприятных условиях среды клетки начинают прикрепляться к подложке (желатиновые подложки) и активно вытягиваться как показано на рисунке 42. Желатиновая подложка является наиболее оптимальным вариантом для работы с культурами клеток фибробластов. В нормальных условиях фибробласты — веретенообразные вытянутые клетки с прозрачной цитоплазмой. Фибробласты обладают ориентированным ростом, появление зернистости в цитоплазме, пустые места в слое клеток, распластанность клеток свидетельствует об их дегенерации в результате токсических или иных вредных воздействий. Небольшое количество среды после каждого пересева отливают для проверки на стерильность.
 Рисунок 42 – Прикрепленные клетки фибробластов
Рисунок 42 – Прикрепленные клетки фибробластов
На 5-7 сутки клетки фибробластов начинают активно делиться, тем самым увеличивая популяцию (рисунок 43).

Рисунок 43 – Культура клеток на 5-7 сутки после пересева
 К 10 — 12 суткам нормальные активированные фибробласты (НАФ) начинают экспрессировать актин гладких мышц (αSMA) и виментин, приобретая звездчатую форму (рисунок 44).
К 10 — 12 суткам нормальные активированные фибробласты (НАФ) начинают экспрессировать актин гладких мышц (αSMA) и виментин, приобретая звездчатую форму (рисунок 44).
Рисунок 44 – Культура клеток на 10-11 сутки после пересева
После образования монослоя клеток, производили второй пересев. В процессе пересева клеток после трипсинизации подсчитывали общее количество клеток, используя камеру Горяева. Пересевы производят для получения количества клеток, достаточного для цитогенетического или биохимического исследования, а также для криоконсервации, то есть для наращивания достаточной культуральной массы. В ходе нашего исследования культура клеток фибробластов использовалась для введения в них генетической конструкции CRISPR.
10 Введение разных вариантов генетических конструкций в фибробласты.
Для доставки генетических конструкций в фибробласты был использован физический трансфекции – электротрансфекция. Данный метод связан с электропорацией — созданием брешей (пор) в мембране клетки. Установлено, что воздействие на клетки импульсов электрического поля высокой напряженности (0,5-15 кВ/см, в зависимости от типа клеток) и различной длительности (10 мкс – 50 мс) ведет к обратимому увеличению проницаемости плазматической мембраны для малых ионов, низко- и высокомолекулярных веществ. Это происходит за счет образования в плазматической мембране пор диаметром 0,5 нм. Несоответствие пор небольшого диаметра и крупных молекул ДНК нивелируется воздействием электрического поля, которое оказывает мощное давление на ДНК и опосредовано на мембрану в области малой поры, расширяя и деформируя ее.
В ходе нашей работы для введения различных вариантов генетических конструкций в фибробласты использовали следующее оборудование и расходные материалы:
1. Культуральные флаконы (матрасы) для культивирования клеток.
2. Чашки Петри (диаметром 5 см) одноразовые
3. Пробирки бактериологические (объемом от 10 до 20 мл)
4. Эппендорф пробирки
5. Пипетки градуированные (пластиковые) (1, 2, 5, 10 мл)
6. Флаконы (пробирки) объемом 10 мл
7. Стерильный сосуд для слива
8. Электропоратор
9. Кюветы для электропоратора
10. Ламинарный шкаф
11. Камера Горяева с покровными стеклами
12. СО2 -инкубатор (термостат) с температурой 37О С
13. Центрифуга на 1000-2000 об /мин
14. Микроскоп инвертированный
Использовали растворы, питательные среды, сыворотки, антибиотики:
— фосфатный буфер (PBS), физ. раствор
— раствор Версена с 0,25% раствором трипсина (1 части трипсина + 3 части версена).
— дезинфицирующие растворы: 70% этиловый спирт
— остовые среды – среда ДМЕМ
— сыворотка крови плодов крупного рогатого скота (ЭТС).
Перед первым использованием разморозить и прогреть при +56ОС — 30 мин или при +45ОС – в течение 1 часа. Рабочее количество хранится при +4ОС, остальное количество при –20ОС.
— глютамин
— нтибиотики – стрептомицин 100 мкг/мл.
-полная среда (с сывороткой) готовится перед ее употреблением и не более чем на 1 месяц работы. Для большинства перевиваемых линий клеток количество сыворотки составляет 5-10 % объема среды роста. Величина рН ростовых сред должна быть в пределах 7,5- 7,6. Все жидкие компоненты, используемые при культивировании, должны быть прозрачными и не иметь осадка. Среду для культивирования фибробластов готовят из стерильной среды ДМЕМ с добавлением 10 % фетальной телячьей сыворотки, стрептомицин 100 мкг/мл.
В нее вносят:
— глютамин 0,3 мг/мл.
— буфер для электропорации ( GTporator)
— реагент P 3000
— ДНК вектор
— липофектамин
Трансфекцию клеток методом электропорации выполняли по следующей схеме:
- Работу вели в ламинарном боксе. Перед работой обрабатывали руки спиртом. Брали чашку Петри с выбранной клеточной культурой.
- Из чашки Петри отбирали 1 мл “старой” среды в пробирку эппендорфа. Центрифугировали, затем отбирали 0,5 мл супернатанта и оставляли, для последующего переноса в новую культуральную посуду.
- Оставшуюся старую среду из ч. Петри утилизировали (сливали) в посуду для отходов. В чашку Петри наливали PBS (с антибиотиком) и аккуратно промывали, сливали в пустую чашку Петри для отходов.
- В чашку Петри с культурой клеток заливали раствор версен + трипсин (2,5 мл) и ставили на 15 минут в термостат (или на термостолик).
Наблюдали под микроскопом за снятием монослоя. Процесс длился 15‑20 минут. - Из чашки Петри раствор с клетками переносили в эппендорф пробирку.
- Центрифугировали пробирку на скорости 2000 оборотов в течение 5 минут. Удаляли надосадочную жидкость.
- Ресуспендировали в 1 мл PBS.
Повторяли пункт 6. - Ресуспендировали в 300 мкл раствора GTporator (буфер для электропорации). Отсюда отбирали 40 мкл раствора с клетками в пробирку эппендорф для подсчета клеток в камере Горяеева.
- Далее раствор центрифугировали. Убирали надосадочную жидкость.
- В отдельную пробирку эппендорф вносили 80 мкл GTporator, добавляли туда 10 мкл плазмидной ДНК, перемешивали.
- В пробирку с открученными клетками (пункт 9) вносили все количество раствора (пункт10), ресуспендировали.
- Собирали все количество раствора и вливали его в кювету.
Кювету вставляли в мультипоратор — нажимали старт. - После этого весь раствор переносили в питательную среду на чашку Петри (или матрас).
- Туда же вносили 0,5 мл старой питательной среды (пункт 2), инкубировали.
Однако этот способ нам не подошел, из-за получения низкого процента трансформированных клеток. И мы решили использовать химический метод трансформации клеток, с помощью липофектамина. Lipofectamine® 3000 Transfection Reagent использует самую современную технологию наночастиц липидов, чтобы обеспечить превосходную эффективность трансфекции с улучшенными результатами применения и воспроизводимыми результатами. Этот реагент обеспечивает превосходную эффективность трансфекции и улучшает жизнеспособность клеток. Реагент Lipofectamine® 3000 предназначен для эффективного трансфекции трудно трансфективных клеток, что дает превосходные показатели трансфекции в широком диапазоне типов клеток. Трансфекцию с применением липофектамина выполняли для культуры клеток, конфлюентность которой составляет 50‑60 %.
Подготовка растворов:
- Смешивали 2 мкл реагента Р 3000 в эппендорф пробирке с 50 мкл среды (без сыворотки и без антибиотика). Пипетировали.
- Содержимое пробирки делили на 2-е эппендорф пробирки по 25 мкл.
- В каждую из 2 эппендорф пробирок добавляли по 2-8 мкл плазмидной ДНК (рассчитываем исходя из концентрации). Пипетировали.
Примечание: Рекомендуемая концентрация вектора 0,8 мкг/мкл (3 мкг на 6 луночный планшет). В пересчете 800 нг/мкл. - Пробирки инкубировали при комнатной температуре 15 минут.
- В это время готовили новый раствор. В новую эппендорф пробирку наливали 50 мкл среды (без антибиотика и без сыворотки) и добавляли 4 мкл липофектамина. Пипетировали.
- После инкубации (шаг 4) в каждую из 2 пробирок добавляли по 25 мкл раствора (среда + липофектамин – шаг 5). Пипетировали.
- Две пробирки инкубировали при комнатной температуре 15 минут.
- После инкубации в каждую пробирку добавляли по 150 мкл среды (без сыворотки и без антибиотика). Получили ДНК-липидный комплекс.
Трансфекцию клеток выполняли следующим образом:
1. Из чашки Петри с культурой клеток убирали старую среду и добавляли приготовленный ДНК-липидный комплекс. Ставили чашку Петри в термостат.
2. Через 3 часа в каждую лунку добавляли по 200 мкл среды с 2-ым содержанием сыворотки (20% сыворотки).
3. Оценку проводили через 2-ое суток.
11 Выращивание культур с модифицированным CRISPR/CAS геномом.
Для культивирования клеток, подвергшихся трансфекции с использованием векторов 111 и 222 использовали следующие расходные материалы и оборудование:
1. Культуральные флаконы (матрасы) для культивирования клеток.
2. Чашки Петри (диаметром 5 см) одноразовые
3. Пробирки бактериологические (объемом от 10 до 20 мл)
4. Пипетки градуированные (пластиковые) (1, 2, 5, 10 мл)
5. Флаконы (пробирки) объемом 10 мл
6. Стерильный сосуд для слива
7. Пинцеты анатомические
8. Камера Горяева с покровными стеклами
9. СО2 -инкубатор (термостат) с температурой 37О С
10. Центрифуга на 1000-2000 об /мин
11. Микроскоп инвертированный
12. Флуоресцентный микроскоп
Использовали растворы, питательные среды, сыворотки, антибиотики:
— фосфатный буфер (PBS), физ. раствор
— раствор Версена с 0,25% раствором трипсина (1 части трипсина + 3 части версена).
— дезинфицирующие растворы: 70% этиловый спирт
— ростовые среды – среда ДМЕМ
— сыворотка крови плодов крупного рогатого скота (ЭТС). Перед первым использованием разморозить и прогреть при +56ОС — 30 мин или при +45ОС – в течение 1 часа. Рабочее количество хранится при +4ОС, остальное количество при –20ОС.
— глютамин
— антибиотики – стрептомицин 100 мкг/мл.
— полная среда (с сывороткой) готовится перед ее употреблением и не более чем на 1 месяц работы. Для большинства перевиваемых линий клеток количество сыворотки составляет 5-10 % объема среды роста. Величина рН ростовых сред должна быть в пределах 7,5- 7,6.
Все жидкие компоненты, используемые при культивировании, должны быть прозрачными и не иметь осадка.
Среду для культивирования фибробластов готовили из стерильной среды ДМЕМ с добавлением 10% фетальной телячьей сыворотки, стрептомицин 100 мкг/мл. В нее вносят: глютамин 0,3 мг/мл.
12 Оценка фенотипа модифицированных клеток.
Дермальные фибробласты играют ключевую роль в физиологии кожи, in vivo характеризуются пластичностью и разнообразием форм – могут иметь овальную, полигональную, веретеновидную или отростчатую форму в зависимости от своего функционального состояния. В культуре дермальные фибробласты обладают способностью прикрепляться к поверхности культуральной посуды и длительно сохранять пролиферативный потенциал.
Оценку культуры модифицированных клеток выполняли на вторые сутки после проведения трансфекции. Культура клеток через двое суток после трансфекции вектором 111 имела конфлюентность 40 %. Культуру фибробластов оценивали в световом микроскопе (Рисунок 45). Культура представлена клетками, прикрепившимися к подложке, вытянутой формы с длинными отростками и круглыми клетками – клетками в активной фазе деления.
 Рисунок 45 –Культура клеток после трансфекции
Рисунок 45 –Культура клеток после трансфекции
Для детекции эксрессии репортерного оранжевого флуоресцентного протеина, закодированного в плазмидном векторе и позволяющем судить об эффективности трансфекции культуры клеток просматривали под люменисцентным микроскопом (рисунок 46). Оранжевый флуоресцентный протеин флуоресцирует при длине волны 600 нм.

Рисунок 46 –Трансфецированная культура клеток под люменисцентным микроскопом
В ходе работы с культурой клеток под флуоресцентным микроскопом нами сделаны некотрые описания фибробластов: клетки веретеновидной уплощенной формы с немногочисленными отростками, с помощью которых образуются межклеточные контакты друг с другом. Ядро в центре, округлое, гипохромное.
Культура клеток, модифицированных с использованием вектора 222 спустя двое суток после трансфекции представлена на рисунке 47. Конфлюентность культуры составила 70 %.
Для детекции эксрессии репортерного оранжевого флуоресцентного протеина, закодированного в плазмидном векторе и позволяющем судить об эффективности трансфекции эту культуру клеток также просматривали под люменисцентным микроскопом при длине волны 600 нм (рисунок 48).
 Рисунок 47 – Культура клеток на 2-е сутки после трансфекции
Рисунок 47 – Культура клеток на 2-е сутки после трансфекции
 Рисунок 48 – Трансфецированная культура клеток с высокой конфлюентностью
Рисунок 48 – Трансфецированная культура клеток с высокой конфлюентностью
На рисунке 49 представлены модифицированные клетки фибробластов веретеновидной формы с немногочисленными отростками при люминисцетной микроскопии.
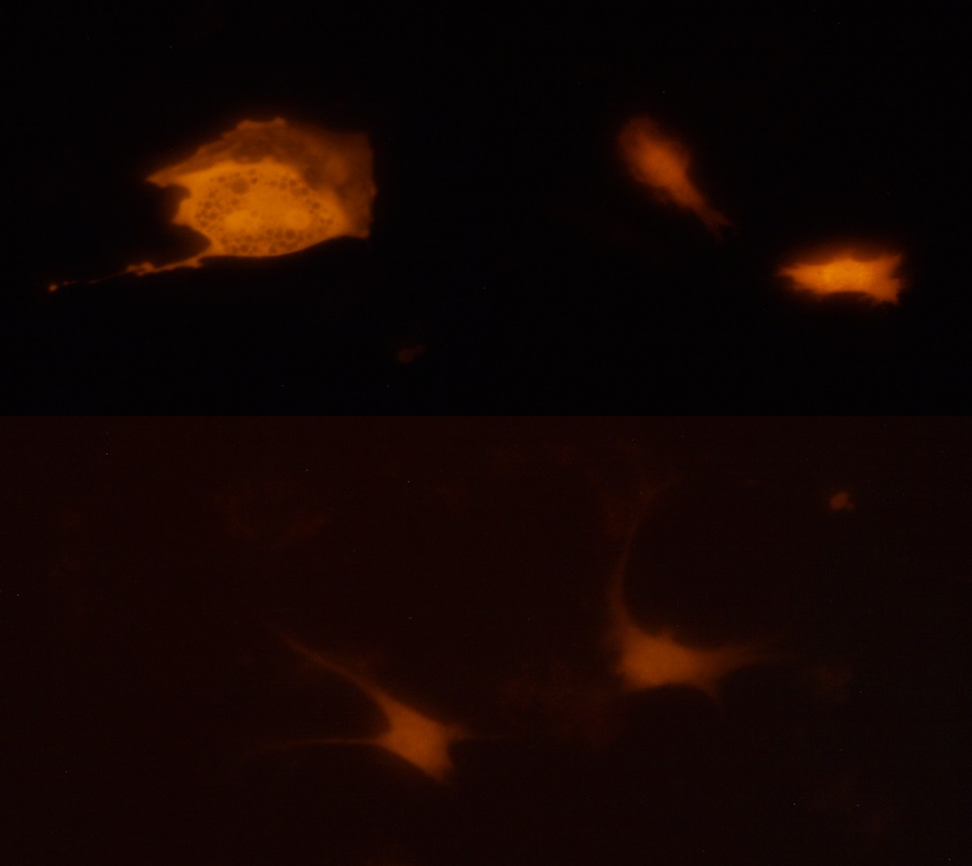 Рисунок 49 – Модифицированные клетки под микроскопом
Рисунок 49 – Модифицированные клетки под микроскопом
На рисунке 50 представлена культура модифицированных клеток при световой микроскопии.

Рисунок 50 – Модифицированные клетки под световым микроскопом
Сравнение морфологии клеток с отредактированным геном с морфологией клеток не подвергающимся модификациям позволяет оценить влияния процесса редактирования на фенотипические характеристики и жизнеспособность культуры фибробластов. В ходе проведенного нами эксперимента каких-либо фенотипических особенностей у модифицированных клеток с отредактированным геномом не обнаружено, что говорит о том, что редактирование не приводит к появлению нецелевых мутаций, затрагивающих гены, участвующие в формировании клеточных структур.
13 Выделение ДНК из фибробластов, оценка эффективности редактирования генов.
После трансфекции оценку результатов клеток проводили через 2 суток, затем, чтобы нарастить культуру перенесли ее в культуральный матрас, поставили в лабораторный инкубатор с атмосферой СО2 на 10 суток. Когда конфлюентность культуры составила 80 %, трансфецированные клетки снимали с подложки методом трипсинизации. Отмывали культуру в фосфатном буфере 2 раза. Готовые пробы были взяты на выделение ДНК.
Выделение ДНК из культуры клеток проводили с использованием набора «QIAamp DNA Mini Kit» производства компании «QIAGEN» (Германия), в специальном ламинарном боксе производитель компания LAMSYSTEMS. Набор предназначен для выделения и очистки общей ДНК (до 50кБ) из цельной и сухой крови, плазмы, сыворотки, лейкоцитов, лимфоцитов, тельных жидкостей, культур клеток, мазков, образцов ткани. Выделение основано на процедуре лизиса клеточных стенок с последующим связыванием ДНК с мембраной (силикой) в миницентрифужной колонке при соответствующей ионной силе и рН. Набор содержит протеиназу К, буферы AL, AW1, AW2 и AE – для элюции ДНК. Основные плюсы набора: быстрая очистка высококачественной ДНК; воспроизводимый и высокий выход ДНК; полное удаление примесей и ингибиторов. Для работы использовали высокоскоростную центрифугу «СМ-50» производитель компания ELMI (Латвия), низкоскоростную центрифугу- вортекс «FVL-2400N Combi-spin» производитель компания BioSan (Латвия), твердотельный термостат «CH‑100» с функцией охлаждения и нагрева производитель компания BioSan (Латвия), прибор Nanophotometer «ImpLEN» для определения концентрации ДНК, а также использовали стерильные пробирки, наконечники, пипетки и дополнительные материалы.
Выделение ДНК проводили согласно протоколу набора «QIAamp DNA Mini Kit». На дно микроцентрифужной пробирки внесли 20 мкл протеиназы К. Добавили в пробирку 200 мкл трансфецированных клеток в фосфатном буфере. Добавили к образцу 200 мкл буфера AL, тщательно перемешали содержимое пробирки с помощью вортекса в течение 15 секунд – это важно для обеспечения эффективного лизиса. Инкубировали пробирку с закрытой крышкой при 56°С в течение 10 минут. Удалили капли с крышки путем кратковременного центрифугирования на вортексе. Добавили 200 мкл этанола 96 % к образцу, снова перемешали с помощью вортекса в течение 15 секунд. Удалили капли с крышки путем кратковременного центрифугирования на вортексе. Осторожно перенесли полученную смесь в спин-колонку QIAamp Mini, избегая при этом смачивания ободка пробирки. Закрыли крышку и центрифугировали пробирку при 8000 об / мин в течение 1 мин. Если после центрифугирования лизат не опустился полностью в собирающую пробирку, необходимо провести повторное центрифугирование на более высокой скорости, до тех пор, пока колонка QIAamp Mini не опустеет. Перенесли спин-колонку QIAamp Mini в чистую собирающую пробирку объемом 2 мл, пробирку, содержащую фильтрат выбросили. Осторожно открыли спин-колонку QIAamp Mini и добавили 500 мкл буфера AW1 избегая при этом смачивания ободка пробирки. Закрыли крышку и центрифугировали пробирки при 13 000 об/мин в течение 1 минуту. Перенесли спин-колонку QIAamp Mini в чистую собирающую пробирку объемом 2 мл, пробирку, содержащую фильтрат выбросили. Осторожно открыли спин-колонку QIAamp Mini и добавили 500 мкл буфера AW2 избегая при этом смачивания ободка пробирки. Закрыли крышку и центрифугировали пробирку на полной скорости (14000 об / мин) в течение 3 минуты. Содержимое собирающей пробирки слили в емкость с отходами. Удалили остатки жидкости с ободка пробирки сухой бумажной салфеткой. Вернули колонку в очищенную собирающую пробирку. Провели дополнительное центрифугирование в течение 1 минуты на максимальной скорости для удаления остатков этанола. Перенесли спин-колонку QIAamp Mini в чистую 1,5 мл микроцентрифужную пробирку. Открыли спин-колонку QIAamp Mini и добавили 200 мкл буфера AE. Инкубировали пробирку при комнатной температуре (23° C) в течение 5 минут, а затем центрифугировали при 8000 об / мин в течение 1 минуты. Извлекли и выбросили колонку из микроцентрифужной пробирки. В микроцентрифужной пробирке находится очищенная ДНК, готовая для дальнейших манипуляций. На приборе «Nanophotometer» проверяли качество, концентрацию ДНК (Рисунок 51).
Рисунок 51 – Концентрация ДНК.
Линзу прибора аккуратно очистили салфеткой, в качестве бланка использовали буфер АЕ (1,5 мкл), в котором была элюирована ДНК. На линзу нанофотометра нанесли 1,5 мкл ДНК. Согласно инструкции, соотношение А260/280 должно быть равным 1,7-1,9 ng/ul.
Соотношение А260/230 имеет второстепенную важность, но должно вписываться в пределы 1,5-3,0 ng/ul (Таблица 16). По данным таблицы видно, что качество выделенной ДНК не высокое. Однако, такой концентрации достаточно для выполнения полимеразной цепной реакции (ПЦР).
Таблица 16 – Проверка качества выделенной ДНК из культур клеток.
| ДНК | Конц. | Ед. | А230 | А260 | А280 | А320 | А260/
А280 |
А260/
А230 |
| Blank (б-р АЕ ) | 0,0000 | ng/ul | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 |
| 11 | 5,3500 | ng/ul | 2,936 | 0,117 | 0,082 | 0,010 | 1,786 | 1,637 |
| 12 | 7,9000 | ng/ul | 8,096 | 0,174 | 0,100 | 0,016 | 1,881 | 1,720 |
| 12 | 6,9500 | ng/ul | 2,689 | 0,155 | 0,110 | 0,016 | 1,879 | 1,952 |
| 21 | 4,5000 | ng/ul | 1,281 | 0,104 | 0,083 | 0,014 | 1,804 | 1,771 |
| 22 | 7,0000 | ng/ul | 1,484 | 0,144 | 0,083 | 0,004 | 1,772 | 1,895 |
| 23 | 7,3000 | ng/ul | 1,717 | 0,159 | 0,109 | 0,013 | 1,721 | 1,986 |
| 24 | 6,7000 | ng/ul | 3,548 | 0,142 | 0,088 | 0,008 | 1,875 | 1,938 |
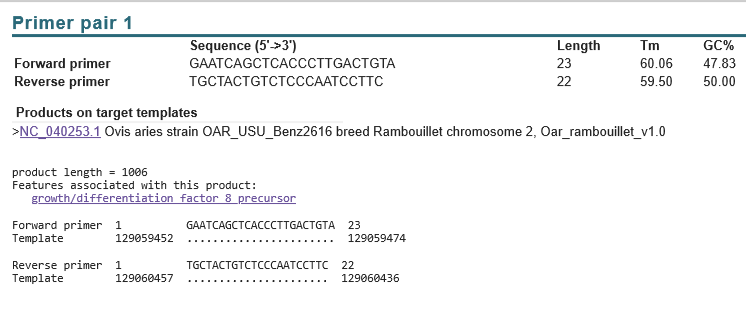 Для амплификации целевого участка были подобраны праймеры, позволяющие охватить область первого экзона гена миостатина овец (Рисунок 52).
Для амплификации целевого участка были подобраны праймеры, позволяющие охватить область первого экзона гена миостатина овец (Рисунок 52).
Рисунок 52 – Праймеры для амплификации экзона 1 гена миостатина овец.
Общая длина ампликона составляет 1006 пар оснований. Расположение целевых последовательностей в ампликоне представлено в таблице 17.
Таблица 17 – Целевые последовательности в ампликоне.
| gRNA | Координаты целевой последовательности в гене | Координаты целевой последовательности в ампликоне |
| Guide1 | 36-53 | 338-355 |
| Guide2 | 112-129 | 414-431 |
Для приготовления ПЦР смеси использовали ПЦР-бокс «Cleaner ПЦР UVC/T-M-AR» производитель компания BioSan. Для обеспечения надежности и быстроты метода детекции локус-специфической рестрикции использовали набор GeneArt® Genomic Cleavage Detection Kit. Набор для определения геномного расщепления GeneArt является быстрым методом, основанный на эндонуклеазе IT7, для количественной оценки редактирования генома клеточной линии (вставки, делеции). Это самый быстрый способ количественной оценки и проверки гРНК CRISPR-Cas9 или нуклеазы TALп. Благодаря набору мы измерили эффективность редактирования генома на мишени, вызванную активностью негомологичного присоединения конца (NHEJ). Преимущества набора GeneArt для определения расщепления генома:
- Минимальное время (ПЦР-амплификация из клеточного лизата, очистка ДНК не требуется);
- Достаточно короткий протокол (четыре часа от отбора клеток до количественных результатов);
- Необходимо просто добавить ПЦР-праймеры — все необходимое в одной коробке, отдельные мастер-миксы для ПЦР или наборы для экстракции ДНК не пригодятся;
- Количественная оценка эффективности редактирования непосредственно с геля. Плотность гелевой полосы напрямую коррелирует с формированием на целевом объекте.
Протокол: клетки диссоциировали TrypLE™ Express, отмыли фосфатно-буферной средой Дульбекко и осадили центрифугированием. Далее клетки подвергали лизису с помощью буфера Cell Lysis Buffer и смеси для белковой деградации Protein Degrader Mix из набора GeneArt® Genomic Cleavage Detection Kit. ДНК экстрагировали и амплифицировали с помощью ПЦР с прямыми и обратными праймерами. Затем проводили реакции денатурации и отжига, чтобы несущие мутации фрагменты отожглись случайным образом с немутированной ДНК. Добавляли детекционный фермент (1 мкл), смесь инкубировали в течение 1 часа при 37°C. Компоненты ПЦР реакции представлены в таблице 18, режим амплификации в таблице 19.
Таблица 18 – Компоненты ПЦР.
| Компонент | Образец (мкл) | Контроль (мкл) |
| Буфер Cell Lysis | 2 | — |
| Смесь праймеров F/R | 1 | — |
| Контрольная матрица | — | 1 |
| AmpliTaq Gold 360 Master Mix | 25 | 25 |
| Вода | 22 | 24 |
| Всего | 50 | 50 |
Таблица 19 – Режим амплификации.
| Этап | Температура | Время | Цикл |
| Активация фермента | 95 °С | 10 мин | 1 |
| Денатурация | 95 °С | 30 сек | 40 |
| Отжиг | 55 °С (Tm) | 30 сек | |
| Элонгация | 72 °С | 30 сек | |
| Финальная элонгация | 72 °С | 7 мин | 1 |
| Хранение | 4 °С | Хранение | 1 |
После завершения амплификации смесь (3 мкл продукта ПЦР с 10 мкл воды) анализировали с помощью электрофореза в 2 % агарозном геле для проверки продукта ПЦР. Для сопоставления размера и количества образца внесли в гель весовой маркер ДНК. На геле выявлены полосы правильного размера — 1000 п.н. Далее был проведен анализ расщепления. В пробирку добавили 2 мкл продукта ПЦР, 1 мкл 10-кратного буфера и воды 6 мкл. Аккуратно центрифугировали. Смесь стала однородной и без пузырьков. Запустили программу ПЦР (Таблица 20).
Таблица 20 – Реакция воссоединения ампликонов.
| Этап | Температура | Время | Температура/время |
| 1 | 95°С | 5 минут | – |
| 2 | 95°С–85°С | – | -2°С/сек |
| 3 | 85°С– 25°С | – | -0,1°С/сек |
| 4 | 4°С | – | Хранение |
На этой стадии гетеродуплексная ДНК, содержащая вставку, делецию расщепляется ферментом обнаружения, что позволяет количественно определить долю геномных модификаций с использованием программы гелевого анализа. В пробирку добавили 1 мкл детектирующего фермента ко всем рестрикцированным образцам, хорошо перемешали. Добавили 1 мкл воды к не рестрикцированным образцам, хорошо перемешали. Инкубировали при 37°C в течение 1 часа, перемешали – 3 секунды, оставили при температуре 4° C. Оценили результаты рестрикции методом E-Gel EX Gel гель-электрофореза. В электрофорезную кювету залили 2% гель E-Gel EX Gel толщиной 0,4 см, осторожно удалили гребенку. В лунки добавили 10 мкл тестируемого образца, в одну лунку добавили контрольный образец. Кювету с гелем поместили в электрофорезную камеру на 30 минут при низком напряжении. Для получения и анализа изображения геля использовали гель-документирующую систему Gel Doc XR+.По результатам гель-документации ампликон полученный в результате амплификации ДНК клеток, отредактированных с использованием вектора 111, после расщепления был представлен тремя полосами-1000 пар оснований (родительская полоса), 650 пар оснований и 350 пар оснований. Ампликон полученный в результате амплификации ДНК клеток, отредактированных с использованием вектора 222 после расщепления, также был представлен тремя полосами-1000 пар оснований (родительская полоса), 550 пар оснований и 650 пар оснований. Полученные результаты говорят о успешно проведенном редактировании первого экзона гена миостатина.
14 Выбор наиболее эффективных вариантов генных конструкций.
В ходе проделанной работы был проведен обширный анализ литературных источников и электронных ресурсов по тематике исследования. Проведенный анализ позволил сконструировать гайдовые последовательности РНК для направленного редактирования первого экзона гена миостатина. С использованием синтезированных специально для эксперимента направляющих нуклеотидных последовательностей и коммерческого линеализированной плазмиды Linearized GeneArt ® CRISPR Nuclease Vector было собрана две различным CRISPR-кассеты (вектор 111, вектор 222). Наработка нуклеазных конструкций проводилась с использованием клонирования в компетентных клетках E. Coli One ShotR. В вектор Linearized GeneArt ® CRISPR Nuclease Vector встроен ген устойчивости к ампицилину, благодаря чему бактерии, успешно пережившие трансформацию и принявшие плазмиды 111 и 222, выжили и в дальнейшем образовали колонии, из которых выделяли плазмидную ДНК. Секвенирование плазмидной ДНК выполнялось для определения направления и последовательности встроенных в вектор нуклеотидов с использованием праймера U6, поставляемого в наборе GeneArt® CRISPR Nuclease Vector Kit. По результатам секвенирования установлено, что участки направляющих последовательностей, выделенные из колоний A222 и В222, трансформированных с использованием вектора 222 имеют правильную ориентацию, нуклеотидный состав и не различаются между собой. В колонии А111, трансформированной с использованием вектора 111 последовательность вектора также не нарушена и имеет правильную ориентацию. Однако вектор, выделенный из колонии В111, трансформированной с использованием вектора 111, содержал делецию TT в области направляющей последовательности и таким, образом не мог быть использован в дальнейшей работе. Для наращивания количества работоспособного вектора 111 и его сохранения в дальнейшей работе использовалась колония A111, для наращивания вектора 222 – колонии А222 и В222. В ходе эксперимента были получены культуры клеток фибробластов от овец северокавказской мясошерстной породы. Введение в фибробласты CRISPR-кассет 111 и 222 осуществлялось методами электропорации и липофекции. Использование электропорации сопровождалось низким выходом жизнеспособных трансформированных клеток. Лучшие результаты были получены с использованием химического метода трансфекции клеток с использованием реактива Lipofectamine® 3000. По результатам наблюдения за трансформированными клетками и оценки фенотипов модифицированных клеток с использованием кассет 111 и 222 установлено, что по своих морфологическим характеристикам клетки подвергшиеся трансформации не отличаются от клеток с неизмененным генотипом, а также друг от друга. Однако, клетки модифицированные с использованием вектора 222 имеют лучшую выживаемость и более активный рост. Так на вторые сутки после трансфекции конфлюентность культуры клеток, модифицированных с использованием плазмиды 222 составляла 70 %, а при использовании плазмиды 111 – только 40 %.
Эффективность редакции также была выше при использовании плазмиды 222. По результатам секвенирования видно, что использованием конструкции 222 чаще сопровождается масштабными инсерциями, а использование 111 – однонуклеотидными заменами (Рисунок 53, 54).
PAM
PAM
–
Рисунок 53 – Выравнивание полученных в результате секвенирования последовательностей экзона 1 гена миостатина
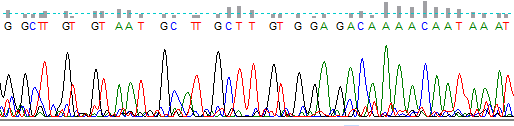
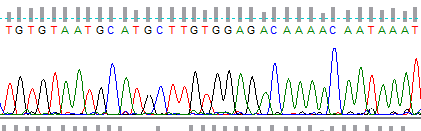
PAM
Рисунок 54 – Хроматограмма секвенирования ДНК клеток с отредактированным геномом
Обобщая полученные данные, с учетом результатов секвенирования используемых векторов, анализа показателей жизнеспособности модифицированных фибробластов и их люминесцентной микроскопии, результатов секвенирования ДНК, выделенных из редактируемых клеток установлено, что обе разработанные CRISPR-кассеты являются работоспособными и пригодны для редактирования клеток овец. Однако, наибольшей эффективностью обладает конструкция 222.
ЗАКЛЮЧЕНИЕ
В ходе проделанной работы проведен анализ научной литературы по вопросам организации структуры генома овец, методам оценки мясной продуктивности овец и существующим модификациям технологии редактирования генома методом CRISPR/Cas с учетом возможности их применения у овец. Подготовлена материально-техническая база для проведения экспериментов по редактированию генома, сконструированы гайдовые последовательности и плазмидные векторы для направленного редактирования первого экзона гена миостатина овец. Получены культуры фибробластов овцы. С использованием методом липофекции и электропорации разработанные генные конструкции внесены в фибробласты овцы. Получены культуры клеток с отредактированным геномом. Модифицированные клетки оценены с использованием световой и люминесцентной микроскопии, а также по результатам секвенирования выделенных ДНК. Разработанные генные конструкции являются работоспособными и пригодны для редактирования клеток овец. Наибольшей эффективностью обладает конструкция 222. В результате работ, выполненных в рамках первого этапа исследований в фибробластах овец отредактирован один из основных маркерных генов мясной продуктивности – ген миостатина. Получены модифицированные монокультуры фибробластов, которые в дальнейшем могут использоваться в качестве ядерных доноров при соматическом клонировании.
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
1. Cong L. еt al. Multiplex genome engineering using CRISPR/Cas systems. // Science — 2013. -Vol. 339, №. 6121. P. 819-823.
2. Mali P. et al. RNA-guided human genome engineering via Cas9. // Science — 2013. — Vol. 339, №. 6121. P. 823-826.
3. Немудрый А.А. и др. Системы редактирования геномов TALEN и CRISPR / Cas – инструменты открытий // 2014. Т. 3. № 22. С. 20–42.
4. Tait-Burkard C. et al. Livestock 2.0 – genome editing for fitter, healthier, and more productive farmed animals // Genome Biol. — 2018. — Vol. 19, № 1. P. 204-215.
5. Hongbing H. et al. One-step generation of myostatin gene knockout sheep via the CRISPR/Cas9 system // Front. Agric. Sci. — 2014. — Vol. 1, № 1. P. 312‑317.
6. Retamales A. et al. Insulin-like growth factor-1 suppresses the Myostatin signaling pathway during myogenic differentiation // Biochem. Biophys. Res. Commun. Academic Press Inc. 2015. — Vol. 464, № 2. P. 596-602.
7. Morissette M.R. et al. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt // Am. J. Physiol. Cell Physiol. 2009. — Vol. 297, № 19. P. 1124-1132.
8. Aiello D. et al. The myostatin gene: an overview of mechanisms of action and its relevance to livestock animals // Anim. Genet. — 2018. — Vol. 3, №5. Р. 449-458.
9. Zhang Y. et al. CRISPR/Cas9‐mediated sheep MSTN gene knockout and promote sSMSCs differentiation // J. Cell. Biochem. John Wiley & Sons, Ltd, 2018. — Vol. 120, № 2. — P. 1794-1806.
10. Wu M. et al. Double-Muscled Phenotype in Mutant Sheep Directed by the CRISPRCas9 System // Cloning Transgenes. — 2018. — Vol. 07, № 01. Р. 312‑320.
11. Wang X. et al. Generation of gene-modified goats targeting MSTN and FGF5 via zygote injection of CRISPR/Cas9 system // Sci. Rep. Nature Publishing Group — 2015. — Vol. 5, № 1. P. 13878-13888.
12. Wang K. et al. CRISPR/Cas9-mediated knockout of myostatin in Chinese indigenous Erhualian pigs // Transgenic Res. Springer International Publishing — 2017. — Vol. 26, № 6. P. 799-805.
13. Lv Q. et al. Efficient Generation of Myostatin Gene Mutated Rabbit by CRISPR/Cas9 // Sci. Rep. Nature Publishing Group — 2016. — Vol. 6, № 1. P. 25029‑25040.
14. Zou Q. et al. Generation of gene-target dogs using CRISPR/Cas9 system // J. Mol. Cell Biol. — 2015. — Vol. 7, № 6. P. 580-583.
15. International Sheep Genomics Consortium et al. The sheep genome reference sequence: a work in progress. // Anim. Genet. — 2010. — Vol. 41, № 5. P. 449-453.
16. Pruitt K.D. et al. RefSeq: an update on mammalian reference sequences // Nucleic Acids Res. — 2014. — Vol. 42, № 1. P. 756-763.
17. Hu X. et al. The complete mitochondrial genome of domestic sheep, Ovis aries // Mitochondrial DNA — 2016. — Vol. 27, № 2. P. 1425-1427.
18. Gorkhali N.A. et al. Mitochondrial DNA variation in indigenous sheep (Ovis aries) breeds of Nepal // Trop. Agric. Res. — 2015. — Vol. 26, № 4. P. 632-641.
19. Zhou H., Hickford J.G.H., Fang Q. Variation in the coding region of the myostatin (GDF8) gene in sheep // Mol. Cell. Probes. — 2008. — Vol. 22, № 1. P. 67‑68.
20. Wang J. et al. Two single nucleotide polymorphisms in the promoter of the ovine myostatin gene (MSTN) and their effect on growth and carcass muscle traits in New Zealand Romney sheep // J. Anim. Breed. Genet. — 2015. – Vol. 4, P. 326-334
21. Sahu A.R. et al. Polymorphism in exon 3 of myostatin (MSTN) gene and its association with growth traits in Indian sheep breeds // Small Rumin. Res. Elsevier — 2017. — Vol. 149, P. 81-84.
22. Pothuraju M. et al. Polymorphism in the coding region sequence of GDF8 Gene in Indian Sheep // Russ. J. Genet. — 2015. — Vol. 51, № 11. P. 1119‑1122.
23. Boman I.A. et al. Impact of two myostatin (MSTN) mutations on weight gain and lamb carcass classification in Norwegian White Sheep (Ovis aries) // Genet. Sel. Evol. — 2010. — Vol. 42, № 1. P. 46-52.
24. Ansary M. et al. Polymorphism in intron-I of myostatin gene and its association with estimated breeding values of growth traits in Baluchi sheep (Ovis aries) // Indian J. Anim. Sci. — 2011. — Vol. 81, № 8. P. 75-78.
25. Clop A. et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. // Nat. Genet. — 2006. — Vol. 38, № 7. P. 813-818.
26. Boman I.A. et al. A frameshift mutation in the coding region of the myostatin gene (MSTN) affects carcass conformation and fatness in Norwegian White Sheep (Ovis aries). // Anim. Genet. — 2009. — Vol. 40, № 4. P. 418-422.
27. Boman I.A., Våge D.I. An insertion in the coding region of the myostatin (MSTN) gene affects carcass conformation and fatness in the Norwegian Spælsau (Ovis aries) // BMC Res. Notes. BioMed Central — 2009. — Vol. 2, № 1. P. 98-112.
28. Dehnavi E. et al. Polymorphism of Myostatin Gene in Intron 1 and 2 and Exon 3, and Their Associations with Yearling Weight, Using PCR-RFLP and PCR-SSCP Techniques in Zel Sheep // Biotechnol. Res. Int. — 2012. — Vol. 2012. P. 111–125.
29. Jeanplong F. et al. Discovery of a mammalian splice variant of myostatin that stimulates myogenesis. // PLoS One. — 2013. — Vol. 8, № 12. P. e81713.
30. Liu C. et al. The critical role of myostatin in differentiation of sheep myoblasts // Biochem. Biophys. Res. Commun. — 2012. — Vol. 422, № 3. P. 381-386.
31. Marcq F. et al. Preliminary results of a whole-genome scan targeting QTL for carcass traits in a Texel x Romanov intercross // 7th World Congr. Genet. Appl. to Livest. Prod. — 2002. — Vol. 22, № 5. P. 282-296.
32. Laville E. et al. Effects of a quantitative trait locus for muscle hypertrophy from Belgian Texel sheep on carcass conformation and muscularity // J. Anim. Sci. — 2004. — Vol. 82, № 11. P. 3128-3139.
33. Beraldi D. et al. Mapping quantitative trait Loci underlying fitness-related traits in a free-living sheep population. // Evolution — 2007. — Vol. 61, № 6. P. 1403-1416.
34. Johnson P.L. et al. Investigations into the GDF8 g+6723G-A polymorphism in New Zealand Texel sheep. // J. Anim. Sci. — 2009. — Vol. 87, № 6. P. 1856-1864.
35. Raadsma H.W. et al. Mapping quantitative trait loci (QTL) in sheep. I. A new male framework linkage map and QTL for growth rate and body weight // Genet. Sel. Evol. BioMed Central — 2009. — Vol. 41, № 1. P. 34-48.
36. Cavanagh C.R. et al. Mapping Quantitative Trait Loci (QTL) in sheep. III. QTL for carcass composition traits derived from CT scans and aligned with a meta-assembly for sheep and cattle carcass QTL. // Genet. Sel. Evol. — 2010. — Vol. 42, № 36. P. 111-124.
37. Johnson P.L. et al. Meat quality traits were unaffected by a quantitative trait locus affecting leg composition traits in Texel sheep. // J. Anim. Sci. — 2005. -Vol. 83, № 12. P. 2729-2735.
38. Gutiérrez-Gil B. et al. Quantitative trait loci underlying milk production traits in sheep // Anim. Genet. — 2009. — Vol. 40, № 4. P. 423-434.
39. Crawford A.M. et al. Discovery of quantitative trait loci for resistance to parasitic nematode infection in sheep: I. Analysis of outcross pedigrees. // BMC Genomics — 2006. — Vol. 7, № 178. P. 115-130.
40. Davies G. et al. Quantitative trait loci associated with parasitic infection in Scottish blackface sheep. // Heredity (Edinb). — 2006. — Vol. 96, № 3. P. 252-258.
41. Телегина Е.Ю. и др. Полиморфизм гена MyoD1 у овец ставропольской породы // Сборник научных трудов Всероссийского Научно-исследовательского института овцеводства и козоводства. 2016. — Т. 2, № 9. С. 254-258.
42. Trukhachev V. et al. The polymorphisms of MyoD1 gene in Manych Merino sheep and its influence on body conformation traits // J. Hell. Vet. Med. Soc.- 2017. — Vol. 68, № 3. P. 319-326.
43. Werf J.H.J. et al. Marker-Assisted Selection in Sheep and Goats // Curr. status Futur. Perspect. Crop. livestock, For. fish. — 2007. — P. 230-247.
44. Álvarez I. et al. Identification of genomic regions and candidate genes of functional importance for gastrointestinal parasite resistance traits in Djallonké sheep of Burkina Faso // Arch. Anim. Breed. — 2019. — Vol. 62, № 1. P. 313-323.
45. Bolotin A. et al. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin // Microbiology -2005. Vol. 151, № 8. P. 2551-2561.
46. Barrangou R. et al. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes // Science — 2007. — Vol. 315, № 5819. P. 1709-1712.
47. Немудрый А.А. Исправление мутации в гене аргинин-вазопрессина крыс линии Brattleboro in vitro // 03.02.07 – генетика Диссертация на соискание ученой степени кандидата биологических наук. ФГБНУ «Федеральный исследовательский центр Институт цитологии и генетики Сибирского отделения Российской академии наук», 2017. С. 1-207.
48. Haft D.H. et al. A Guild of 45 CRISPR-Associated (Cas) Protein Families and Multiple CRISPR/Cas Subtypes Exist in Prokaryotic Genomes // PLoS Comput. Biol. — 2005. — Vol. 1, № 6. P. 60-72.
49. Jiang S. et al. Principles of gene editing techniques and applications in animal husbandry // 3 Biotech. Springer International Publishing — 2019. — Vol. 9, № 1. P. 31-49.
50. Deltcheva E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III // Nature. — 2011. — Vol. 471, № 7340. P. 602-607.
51. Karvelis T. et al. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus // RNA Biol. — 2013. — Vol. 10, № 5. P. 841-851.
52. Anders C. et al. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease // Nature — 2014. — Vol. 513, № 7519. P. 569‑573.
53. Sternberg S.H. et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9 // Nature. — 2014. — Vol. 507, № 7490. P. 62-67.
54. Ruan J. et al. Genome editing in livestock: Are we ready for a revolution in animal breeding industry // Transgenic Res. Springer International Publishing — 2017. — Vol. 26, № 6. P. 715-726.
55. Ma X. et al. In vivo genome editing thrives with diversified CRISPR technologies // Zool. Res. — 2018. — Vol. 39, № 2. P. 58-71.
56. Butler J.R., Tector A.J. CRISPR genome-editing: A medical revolution // J. Thorac. Cardiovasc. Surg. Elsevier — 2017. — Vol. 153, № 2. P. 488-491.
57. Li K. et al. Optimization of Genome Engineering Approaches with the CRISPR/Cas9 System // PLoS One — 2014. — Vol. 9, № 8. P. e105779.
58. Proudfoot C. et al. Genome edited sheep and cattle. // Transgenic Res. — 2015. — Vol. 24, № 1. P. 147-153.
59. Kalds P. et al. Sheep and goat genome engineering: From random transgenesis to the CRISPR era // Front. Genet. — 2019. — Vol. 10, № 2. P. 21-37.
60. Ishii T. Genome-edited livestock: Ethics and social acceptance // Anim. Front. — 2017. — Vol. 7, № 2. P. 24-35.
61. Van Eenennaam A.L. Genetic modification of food animals // Curr. Opin. Biotechnol. Elsevier Current Trends — 2017. — Vol. 44. P. 27-34.
62. Su X. et al. Efficient genome editing in cultured cells and embryos of Debao pig and swamp buffalo using the CRISPR/Cas9 system // Vitr. Cell. Dev. Biol. — Anim. In Vitro Cellular & Developmental Biology – Animal — 2018. — Vol. 54, № 5. P. 375-383.
63. Hai T. et al. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system // Cell Res. — 2014. — Vol. 24, № 3. P. 372-375.
64. Zhang T. Production of transgenic swine by sperm stem cell transplantation and gene editing technology // AIP Conf. Proc. — 2019. — Vol. 28, № 2. Р.612-625.
65. Валетдинова К.Р. Применение системы CRISPR/Cas9 для создания и исследования клеточных моделей наследственных заболеваний человека // Наука о генетике — 2016. — №2. P. 10-20.
66. Crispo M. et al. Efficient Generation of Myostatin Knock-Out Sheep Using CRISPR/Cas9 Technology and Microinjection into Zygotes // PLoS One / ed. Veitia R.A. IETS — 2015. — Vol. 10, № 8. P. e0136690.
67. Zhou W. et al. Generation of beta-lactoglobulin knock-out goats using CRISPR/Cas9 // PLoS One / ed. Sun Q.-Y. Public Library of Science — 2017. Vol. 12, № 10. P. e0186056.
68. Zhang J. et al. Comparison of gene editing efficiencies of CRISPR/Cas9 and TALEN for generation of MSTN knock-out cashmere goats // Theriogenology. Elsevier Ltd — 2019. — Vol. 132, P. 61-71.
69. Yan Q. et al. Generation of multi-gene knockout rabbits using the Cas9/gRNA system // Cell Regen. BioMed Central — 2014. — Vol. 3, № 1. P. 3-12.
70. Гридина М.М. Способы повышения эффективности knock- in в геном плюрипотентных клеток человека при помощи системы CRISPR/Cas9 // Vavilov J. Genet. Breed. — 2019. — Vol. 22, № 8. P. 1026-1032.
71. Zhang J. et al. CRISPR/Cas9-mediated specific integration of fat-1 at the goat MSTN locus // FEBS J. — 2018. — Vol. 285, № 15. P. 2828-2839.
72. Bi Y. et al. Isozygous and selectable marker-free MSTN knockout cloned pigs generated by the combined use of CRISPR/Cas9 and Cre/LoxP // Sci. Rep. Nature Publishing Group — 2016. — Vol. 6, № 1. P. 31729-31738.
73. Stinckens A. et al. Mutations in the Myostatin gene leading to hypermuscularity in mammals: Indications for a similar mechanism in fish // Animal Genetics — 2011. — Vol. 42, № 3. P. 229-234.
74. Ahad W.A. et al. Applications of Myostatin (MSTN) Gene in the Livestock Animals and Humans : A Review // Int. J. Curr. Microbiol. App. Sci. — 2017. — Vol. 6, № 9. P. 1807-1811.
75. Li J. et al. Regulation of myostatin promoter activity by myocyte enhancer factor 2 // Sheng Wu Gong Cheng Xue Bao — 2012. — Vol. 28, № 8. P. 918‑926.
76. Stefaniuk M. et al. MSTN gene polymorphism in livestock animals // Postepy Hig. Med. Dosw. — 2014. — Vol. 68. P. 633-639.
77. Invitrogen. GeneArt ® CRISPR Nuclease Vector Kit // Manual. — 2013. — № 7. P. 9-16.
78. Данлыбаева Г.А. и др. Технология получения и культивирования аллогенных и аутологичных фибробластов человека для заместительной клеточной терапии // Биотехнология. Теория и практика. – 2011. — № 2. С. 77 ‑82.
79. Беляев, В.А. и др. Исследование возможности ведения фибробластов в перевиваемых культурах // Вестник АПК Ставрополья. – 2015. — №1 (17). С. 80-84.
80. Витрук Г.А. и др. Дермальные фибробласты и старение кожи человека// Медицина – 2003. №6. С. 31-37.