
Титульный лист и исполнители
Отчет 75 с., 36 рис., 20 табл., 64 источн.
CRISPR, CAS9, MSTN, МИОСТАТИН, ОВЦА, ГЕНОМНОЕ РЕДАКТИРОВАНИЕ, ФИБРОБЛАСТ, ГЕН, МИКРОСКОПИЯ, СЕКВЕНИРОВАНИЕ
В отчете представлена информация о современных технологиях направленного геномного редактирования с использованием системы CRISPR/Cas9, для целевого трансгенеза и внесения новых последовательностей в геном эукариот, полученная при изучении источников иностранной литературы.В ходе работы подготовлена материально-техническая база для проведения экспериментов по редактированию генома, сконструированы редактирующие векторы и донорные последовательности для направленного редактирования первого экзона гена миостатина овец. Проведен компьютерный анализ и конструирование гидовых последовательностей РНК и донорных последовательностей. Получены олигонуклеотиды и плазмидные векторов, кодирующие систему CRISPR/Cas9. Произведена наработка редактирующих векторов путем клонирования в компетентных клетках E. Coli One ShotR с последующей очисткой. Получена культура клеток аутологичных дермальных фибробластов овцы.Проведена сравнительная оценка эффективности внесения разработанных редактирующих конструкций в фибробласты овцы с использованием физического и биохимического метода трансфекции. Произведена оценка фенотипа модифицированных клеток. Проведена оценка эффективности геномного редактирования, определены наиболее эффективные варианты генных конструкций и их доставки . Согласно полученным данным, и с учетом эффективности геномного редактировании при использовании различных векторов, наиболее эффективным вариантом генных конструкций для нокаута гена миостатина при негомологичного соединении концов являются векторы CRISPR3 и CRISPR38. Для запуска механизма гомологичной рекомбинации с целью интеграции добавочных стоп кодонов наиболее предпочтительным является использование редактирующего вектора CRISPR3в сочетании с донорной последовательностью ssODN3.Наиболее эффективным способом доставки используемых редактирующих конструкций в фибробласты овец, согласно проведенным исследованиям, является метод биохимической трансфекции – липофекции. Эффективность трансфекции фибробластов с использованием метода липофекции и реагента Lipofectamine 3000 была на 16 % выше, чем с использованием метода электропорации и составила 28 %.
Введение
Развитие геномных технологий на фоне возрастающей экономической значимости мясной продуктивности сельскохозяйственных животных обуславливает перспективы получения новых селекционных форм с использованием методов геномного редактирования. Решение вопросов направленного геномного редактирования генов, функции которых напрямую связаны с ростом и развитием организма млекопитающих, является актуальным как для мировой фундаментальной генетики, так и для прикладных аспектов селекции сельскохозяйственных животных. Так редактирование последовательности гена миостатина, ограничивающего рост мышечной ткани, позволяет получить животных, с хорошо развитой мускулатурой. Овцы с отредактированным геном миостатина превосходят по живой массе животных контрольной группы в среднем на 29 %, козы – на 32 %, свиньи – на 14 %.
Система CRISPR/Cas9 (clustered regulatory interspaced short palindromic repeats; короткие палиндромные повторы, расположенные группами, равномерно удаленными друг от друга) на сегодняшний день является одной из наиболее востребованных и эффективных редактирующих систем [1–3]. Внесение изменений в целевой сайт обеспечивается комплексом единой химерной направляющей/гидовой РНК и крупного полифункционального белка – Cas9, что обеспечивает ее относительно низкую стоимость и простоту [4–6]. Подбор целевой последовательности при этом ограничен необходимостью присутствия на 3’-конце мотива PAM (protospacer-adjacentmotif, мотив, прилегающий к протоспейсеру) состоящего из 3 нуклеотидов NGG[7–9].
Одним из лидеров в области CRISPR/Cas редактирования является Китай. Именно в Китае в 2014 году с применением системы CRISPR/Cas9 были получены первые клинически здоровые ягнята с отредактированной последовательностью гена миостатина (GDF-8, MSTN, TGF-8) – одного из ключевых регуляторов мышечного роста, ограничивающего дифференцировку и пролиферацию миосателлитов, миобластов и некоторых других видов клеток [10–13]. К 2018 году с использованием системы CRISPR/Cas9 учеными из Китая были получены первые ягнята нуль-мутанты по гену миостатина обладающие фенотипом двойной мускулатуры [14,15]. Аналогичные эксперименты проводятся на козах, свиньях и животных других видов по всему миру [16–19].
В связи с высокой актуальностью вопросов получения CRISPR/Cas9 модифицированных клеточных линий, эмбрионов и животных, обусловленной не только общемировым научным интересом, но и перспективами практического внедрения технологии в сельское хозяйство, целью наших исследований явилась разработка методов использования технологии редактирования генома CRISPR/CAS для повышения продуктивности сельскохозяйственных животных на примере овец российских пород.
1. Изучение источников иностранной литературы с информацией о современных технологиях направленного геномного редактирования с использованием системы CRISPR/Cas9
Система CRISPR/Cas9 – наиболее широко используемый метод введения в ДНК сайт-специфичных двуцепочечных разрывов (DSB). Свою популярность он заслужил благодаря высокой специфичности и эффективности, сопряженным с простотой исполнения и надежностью (Hsu et al., 2014). [1–3]. Внесение изменений в целевой сайт обеспечивается комплексом единой химерной направляющей/гидовой РНК(single guide RNA,sgRNA) и крупного полифункционального белка – Cas9[20].
Cas9 – бактериальная РНК-управляемая эндонуклеаза, использующая спаривание оснований для распознавания и расщепления целевых ДНК комплементарных направляющей РНК.Белок Cas9, имеет шесть доменов: REC I, REC II, Bridge Helix, PAM Interacting, HNH и RuvC. Самый крупный домен Rec Iотвечает за связывание направляющей РНК. Богатый аргинином домен «мостиковая спираль» (Bridge Helix) необходим для инициации расщепляющей активности связывания целевой ДНК. Домен, взаимодействующий с PAM (PAM Interacting), отвечает за инициирование расщепления целевой ДНК. Домены HNH и RuvC – нуклеазные домены, разрезающиецепи ДНК, формируя DSB.При этомHNH домен отвечает заразрезаниепоследовательности-мишени, комплементарной sgRNA, а домен RuvC отвечает за расщепление нецелевой последовательности.ВотсутствиенаправляющейРНКбелокCas9 остаетсянеактивным. Связывание направляющей РНК и белка Cas9 вызывает конформационное изменение белка, что приводит к его активации[21]. В генной инженерии используется химерная sgRNA, вторичная структура которой имитирует комплекс зрелой crРНК и tracrРНК за счет структуры стебель-петля (повтор-антиповтор в комплексе crРНК-tracrРНК). С инженерной точки зрения, sgRNA состоит из двух частей: константной части, которая образуется из нескольких петель и связывает Cas9, и 5′-концевой короткой вариабельной спейсерной части, состоящей из 20 нуклеотидов.
Система CRISPR/Cas9получила широкое применение при редактировании генов, влияющих на формирование продуктивных признаков у сельскохозяйственных животных. Особый интерес для исследователей представляло изменение последовательности гена миостатина (MSTN, GDF-8, TGF-8), ограничивающего дифференцировку и пролиферацию мышечных клеток[12,13,22]. Установлено, чтонокаутгенамиостатинапозволяетполучитьживотных, отличающихся интенсивным развитием мышечной ткани и высокой мясной продуктивностью. Овцы с отредактированным геном миостатина превосходят по живой массе животных контрольной группы в среднем на 29 %, козы – на 32 %, свиньи – на 14 % [18,23–25]. При получении сельскохозяйственных животных с отредактированным геном миостатина, система CRISPR/Cas9использовалась в 16 экспериментах из 24. Редактирование гена выполнялось как путем внесения случайных делеций и инсерций, так и путем внесения заданных последовательностей с использованием донорных олигонуклеотидов при гомологичной рекомбинации. В область гена вносили точечные мутации, интегрировали добавочные стоп-кодоны и целые генные последовательности [26–28]. При редактировании генома эмбрионов ученые столкнулись с проблемой рождения генетических мозаиков. Редактирующая система зачастую по-разному срабатывала в отдельных клетках развивающегося организма, что приводило к появлению большого количества различных мутантных аллелей [23]. Решением стало использование технологии переноса ядер соматических клеток. Соматическое клонирование клеток с отредактированным геном при тщательном отборе донорских клеток в большинстве случаев позволяло избежать рождения особей с нежелательным генотипом [29]. Нокаут эндогенного миостатина при сайт-специфичном нокине с использованием генных последовательностей, задействованных в синтезе биологически активных веществ, может быть использован не только для увеличения мышечной массы сельскохозяйственных животных, но и для насыщения получаемого мяса ценными химическими составляющими[30]. В работах, проведенных на козах и свиньях, было показано, что животные с отредактированным геном миостатина способны передавать по наследству мутантные аллели [31,32].
Получение овец с отредактированным геном MSTN.При получении овец с отредактированным геном MSTN с использованием системы CRISPR/Cas9 редактирование преимущественно проводилось на стадии зиготы. Готовые в vitro синтезированные мРНК Cas9 и единую химерную направляющую РНК (хнРНК) вносили в эмбрионы путем микроинъекций, затем выжившие эмбрионы трансфицировали самкам-реципиентам. В 2014 году впервые получено два разнополых ягненка с измененной последовательностью третьего экзона, животные являлись генетическими мозаиками. У самки наряду с аллелем дикого типа выявлено два мутантных аллеля, у самца – пять. Только три обнаруженных мутации приводили к сдвигу рамки считывания. Рождаемость носителей мутантных аллелей составил менее 1 %. Из 213 трансфицированных эмбрионов родилось 35 ягнят, ожидаемые мутации несли только 2 ягненка[10].
В 2015 году получены клинически здоровые гомозиготные нуль-мутанты по гену MSTN с отредактированной последовательностью первого экзона.Из 216 зигот до стадии бластоцисты развились 53, которые были трансфицированы реципиентам. Родилось 19 клинически здоровых ягнят, 8 из которых несли мутантные аллели в разных формах и комбинациях (инсерции и делеции длиной от 2 до 25 пар оснований). Формирование преждевременных стоп-кодонов после внесения мутаций обнаружено у 7 животных, 4 из них являлись гомозиготными нуль-мутантами по гену MSTN. Нуль-мутанты в эмбриональном периоде и при рождении не отличались по своим характеристикам от животных с диким генотипом. Однако уже на 15 день жизни были выявлены достоверные различия по живой массе, которые на 60 день составляли 20-30 %. Ягнята с одной неработоспособной копией гена не имели достоверных различий по живой массе в сравнении с животными дикого типа [23].
В 2018 году получены клинически здоровые овцы с фенотипом двойной мускулатуры. Из 32 рожденных ягнят, только 5 несли мутантные аллели. Редактирование первого экзона привело к появлению инсерций и делеций различной длины. Живая масса всех носителей мутантных аллелей при рождении была достоверно выше, чем у животных с диким генотипом, однако фенотип двойной мускулатуры проявился только у 2 ягнят из 5. К формированию фенотипа двойной мускулатуры, сопровождающегося гипертрофией и гиперплазией мышечных волокон в сочетании с резким снижением уровня экспрессии миостатина привели делеции длиной в 1 и 2 п.о.[15].
В 2019 году получено 10 клинически здоровых ягнят с отредактированной последовательностью гена MSTN. В зиготы одновременно инъецировали три различных хнРНК, запрограмированных на узнавание 1, 2 и 3 экзонов. Такой подход позволил добиться внесения крупных делеций, длиной до 5 т.п.н. Однако, полученные животные были либо гетерозиготами, либо мозаиками. Носители мутаций достоверно превосходили животных дикого генотипа по среднесуточному приросту [33].
Систему CRISPR/Cas9 также использовали при получении ягнят-клонов с нокаутированным MSTN. Редактирование гена было проведено в фетальных фибробластах, мутантные клетки имели в области второго экзона делецию длиной 21 п.н. Путем переноса ядер соматических клеток (Somatic Cell Nuclear Transfer, SCNT) фибробластов с отредактированной последовательностью гена MSTN в энуклеированные ооциты были получены клоны с гистологически подтвержденной гипертрофией мышечных волокон[14].
Получение коз с отредактированным геном MSTN. Основным способом получения мутантных эмбрионов коз являлось реконструирование с использованием SCNT. Первые клонированные козлята нуль-мутанты по гену MSTN получены в 2014 году. Геном донорных MSTN-KO фибробластов, был отредактирован системой CRISPR/Cas9 в область второго экзона внесены биаллельные делеции длинной от 2 до 11 п.н. Процедуры SCNT и эмбриотрансфер выполнялись по стандартным протоколам. В общей сложности самкам-реципиентам был подсажен 21 эмбрион, наступило 7 беременностей. В результате эксперимента получено 3 козленка с генотипом идентичным генотипу донорской клетки. Один козленок умер вскоре после рождения, что авторы статьи связывают с последствиями SCNT. Оставшиеся в живых козы были клинически здоровы. При проведении вестерн-блот анализа миостатин в мышцах мутантных животных не обнаруживался [29].
В 2018 году опубликованы результаты работы по получению клонированных коз с одновременным нокаутом эндогенного MSTN и сайт-специфичном нокине экзогенного fat-1. Эффективность CRISPR/Cas9 опосредованного генного нокаута и направленной интеграции гена fat-1 в область первого экзона гена MSTN составила 25,6 %. Полученные мутантные клеточные линии были гетерозиготны по желаемой модификации. С использованием технологии SCNT получен 1 трансгенный козленок. Уровень экспрессии гена fat-1 у трансгенного животного был в 1,5 раза выше, чем у животных дикого типа, в то время как уровень экспрессии MSTN практически вполовину ниже. Не смотря на высокий уровень экспрессии гена fat-1, содержание ПНЖК n-6 в клетках мутантного животного было значительно ниже, чем в клетках животных контрольной группы, содержание кислот n-3 достоверно не различалось [34].
Сравнительное изучение эффективности систем TALEN и CRISPR/Cas9 при получении коз с отредактированным геном MSTN проведено в 2019 году. Эффективность внесения биаллельных мутаций с использованием системы CRISPR/Cas9 на клеточном уровне была в 8,5 раз выше, чем с использованием системы TALEN, также большей была максимальная длина вносимых делеций – 117 и 13 п.н. соответственно. Живую MSTN-KO козу удалось получить только при использовании в качестве ядерных доноров, клеток, отредактированных системой CRISPR/Cas9. Уровень экспрессии MSTN в 6-ти месячном возрасте у носителя мутации был на 50 % ниже, чем у животных контрольной группы [35].
В двух экспериментах получение коз с отредактированной последовательностью гена MSTN осуществлялось путем микроинъекций мРНК Cas9 и хнРНК в зиготы. В 2015 году R. Guo с соавторами получили клинически здоровую козу с измененной последовательностью экзона 3, имеющую короткие инсерции и делеции длиной от 1 до 3 п.н. [36]
X. Wang с соавторами для редактирования последовательности гена использовали РНК, запрограммированные на узнавание 2 и 3 экзонов. Зиготы для последующего редактирования вымывались из яйцеводов самок через 14 часов после спаривания. Выбранная методика была довольно трудоемкой и имела низкую эффективность. Так в результате выполнения микроинъекции из 862 используемых зигот погибло 446, и только 15 из 79 новорожденных ягнят несли мутантные аллели гена MSTN (делеции и инсерции длиной от 1 до 23 п.н.)[16]. За животными наблюдали в течение 360 дней. На протяжении этого периода, в том числе при рождении и отъеме, носители мутаций достоверно превосходили по живой массе животных контрольной группы. По данным лабораторных исследований биохимические показатели крови у мутантных животных на 120 день жизни находились в пределах нормы, достоверных отличий от показателей животных контрольной группы выявлено не было. По результатам ПЦР и вестерн-блотт анализа уровень экспрессии гена MSTN у носителей мутации был значительно ниже, чем у животных контрольной группы. Установлено, что мутантные особи были способны к размножению и передавали по наследству имеющиеся мутации [32].
Получение свиней с отредактированным геном MSTN. В качестве основного метода получения эмбрионов свиньи с отредактированным геном MSTNпри использовании системы CRISPR/Cas9преимущественно применялся метод SCNT. Так в 2015 году было получено 8 мутантных поросят-клонов с отредактированной последовательность третьего экзона гена MSTN: два мертворожденных и шесть умерших в течение первой недели после рождения. Дляредактированиябылииспользованыфетальныефибробластысвинейпородыландрасикитайскойпородыэрхуалиан (Erhualian).Длятрансфекциииспользовалидваразличныхвекторанаправленныхнаредактированиеэкзона 3 генамиостатина, отличающихсяпоследовательностяминаправляющейРНК. Эффективностьредакцииприиспользованиивекторовпоотдельностинепревышала 21.79 %, прико-трансфекции двух различных векторов эффективность составляла 31,5 %. Интересным является то, что в данной работе на фибробластах свиньи была продемонстрирована возможность использования вспомогательных матричных одноцепочечных олигонуклеотидов для запуска опосредованной гомологичной рекомбинация при ко-трансфекции с нуклеазным вектором, однако эффективность гомологичной рекомбинации для внесения ожидаемой инсерции составила только 3,64 %. Из 51 колонии фибробластов, полученной от свиней породы ландрас и прошедшей процедуру трансфекции 10 колоний несли интересующие мутации, из 37 колоний полученных от эрхуальских свиней – мутации несли 13 колоний. В качестве донорских клеток для SCNT были выбраны четыре колонии фибробластов предположительных нуль-мутантов по гену миостатина, полученных от свиней породы ландрас. С использованием технологии SCNT было получено 8 мутантных поросят-клонов: два мертворожденных и шесть умерших в течение первой недели после рождения. Живая масса при рождении у мутантных особей в среднем была на 15% больше, чем у однопометников с диким генотиипом.По результатам гистологических исследований у мутантов обнаружена гиперплазия мышечных волокон. Уровень экспрессии MSTN в сердечной мышце мутантных поросят был в разы ниже, чем в мышце поросят контрольной группы, в то время как уровень экспрессии MSTN в фибробластах значительно не различался[37].
В 2016 году мутантных клонов получили с использованием CRISPR/Cas9 опосредованной гомологичной рекомбинации и системы Cre/LoxP. Вектор CRISPR/Cas9 вводили в фетальные фибробласты свиньи совместно с донорской ДНК, включающей в себя последовательности, гомологичные экзону 3 гена MSTN, с удаленным участком, кодирующим С-концевой фрагмент зрелой формы миостатина (172 п.н.). Также вносили последовательность селективного маркерного гена зеленого флуоресцирующего протеина (green fluorescent protein, GFP) флоксированную LoxP сайтами. Безошибочная интеграция донорной последовательности была выявлена только у 1 клеточной линии, причем в гетерозиготном состоянии. Эффективность гомологичной рекомбинации составила 7,7 %. Далее флоксированный ген GFP был удален с использованием Cre-рекомбиназы. Фибробласты с отредактированной последовательностью MSTN и с удаленным GFP использовались в качестве ядерных доноров при SCNT. Получено 2 здоровых поросенка (самцы), с генотипом идентичным генотипам клеток-доноров. Экспрессия MSTN в мышцах мутантных клонов была на 50% ниже по сравнению с животными контрольной группы при усиленной экспрессии генов MyoD1, MyoG и MYF5. Гистологический анализ показал, что нокаут MSTN вызывал мышечную гиперплазию. В первые три месяца жизни достоверных различий по живой массе между животными не наблюдалось, однако, в более старшем возрасте поросята MSTN KO достоверно превосходили по живой массе поросят контрольной группы на 12 % [38].
В 2017 году для получения клонов нуль-мутантов по гену MSTN использовали фибробласты с измененной последовательностью третьего экзона, имеющие делецию (104 п.н.) и единичную инсерцию (1 п.н). После SCNT было получено 26 свиней-клонов, 23 из них были гомозиготными по ожидаемым мутациям. Средняя масса поросят с генотипом MSTN-/- при рождении достоверно не отличалась от поросят дикого генотипа. В возрасте 1 месяца проявились ярко выраженные фенотипические различия в виде более широкой спины и хорошо развитых мышц. У MSTN-/- поросят резко снизился уровень белка-предшественника миостатина. Внесенные мутации привели к появлению добавочного стоп-кодона в экзоне 3 и утрате его функциональной активности [39].
В 2019 году для получения поросят-клонов мутантных по гену MSTN в качестве ядерных доноров при SCNT впервые использовали клетки почки. Рекомбинантную плазмиду CRISPR/Cas9, запрограммированную на редактирование первого экзона доставляли в клетки путем электропорации. В результате проведенной работы было получено 3 мутантных клона, однако, в связи с погрешностями отбора донорских клеток, все они имели разные генотипы. Полученные животные несли мутантных аллели трех типов с делециями длинной 4 и 6 п.н..Гистологически отмечалось увеличение числа мышечных волокон при одновременном уменьшении их размера. Все мутантные особи пали от энзоотической пневмонии свиней в возрасте 18-120 дней [40].
Первое успешное редактирование гена MSTN в зиготах свиней с последующим получением клинически здоровых поросят проведено в 2016 году. Белок Cas9 и хнРНК вводили в зиготы путем электропорации. Мутации в гене MSTN несли 9 из 10 полученных свиней, однако у большинства из них обнаружились и аллели дикого типа. Единственный нуль мутант по гену MSTN был мозаиком, несущим три мутантных аллеля, связанных со сдвигом рамки считывания и отличался более выраженной мускулатурой. На 40 день жизни белок миостатина в его мышцах не обнаруживался. Самцы с отредактированным геном были способны к размножению, при этом внесенные мутации передавались по наследству [41].
В 2018 году при работе с эмбрионами свиней для редактирования гена MSTN использовали микроинъекции мРНК Cas9 и хнРНК. Эффективность редактирования генома эмбрионов составила от 14,3 до 36,4 %. В последовательность гена MSTN выли внесены инсерции и делеции длиной от 1 до 45 п.н., при этом наблюдался мозаицизм [42].
Система CRISPR/Cas9 на сегодняшний день является наиболее востребованным инструментом, позволяющих решать задачи высокоточного редактирования генома сельскохозяйственных животных. Для доставки редактирующих конструкций при работе с соматическими клетками чаще используется методы электропорации и липофекции, при работе с половыми клетками и эмбрионами микроинъекция и электропорация. С развитием технологий редактирование движется от простого внесения случайных делеций и инсерций при негомологичном соединении концов к интеграции длинных генных последовательностей путем запуска гомологичной рекомбинации. Перспективным является направление нокаута нокином с использованием генных последовательностей маркерных генов, или генов, задействованных в синтезе ценных биологически активных веществ.
2. Анализ мирового опыта использования системы CRISPR/Cas9 для направленного трансгенеза и внесения новых последовательностей в геном эукариот с использованием плазмидных CRISPR/CAS кассет и дополнительных ДНК-матриц
В настоящее время система CRISPR/Cas9 широко используется для введения в ДНК сайт-специфичных двуцепочечных разрывов (double strand breaks, DSB).С ее помощью с высокой эффективностью удается реализовать нокаут (knock-out) интересующих исследователя генов. Однако введение в целевое место генома заданной последовательности – нокин (knock-in) является существенно более сложной задачей[20].
Репарацию DSBклетки осуществляют,используя механизмы негомологичного соединения концов (nonhomologous end joining, NHEJ) или механизмы гомологичной рекомбинации (homology-directed repair, HDR) [43]. NHEJ – это неспецифическая реакция лигирования, точность которой сильно зависит от структуры концов разрыва, а результатом могут быть различные инсерции или делеции (инделы) в целевой участок генома. При использовании NHEJ получают нокаут интересующих генов.Для получения целевой встройки в геном при запуске механизма гомологичной рекомбинации вместе с компонентами CRISPR/Cas9-системы в клетку вносят матричную последовательность с участками гомологии по обеим сторонам от DSB, которая будет служить донором при репарации[20]. В качестве донорной последовательности используется плазмидная двуцепочечная ДНК (double stranded DNA,dsDNA) или синтетический одноцепочечный олигонуклеотид (single-stranded oligonucleotide,ssODN). Плазмидная ДНК позволяет встраивать в геном достаточно крупные фрагменты – до 7.4 т. п. о. [44]. В случае необходимости встройки короткой последовательности, оправдано использование ssODN. Он позволяет с более высокой эффективностью, по сравнению с использованием dsDNA, вводить последовательности размером до 100 нуклеотидов[45,46]. Это особо привлекательно в свете возможности проведения корректировки точковых мутаций [47,48].
Для увеличения эффективности направленного нокина разработано несколько основных стратегий подбора донорных последовательностей, связанных с варьированием длины плеч гомологии и комплементарностью используемых матриц.Типичным дизайном ssODN являются симметричные плечи гомологии, соответствующие последовательностям по обе стороны от внесенного разрыва. По данным Liangи соавторов увеличить эффективность гомологичной рекомбинации позволяет использование асимметричного ssODN, у которого 36 нуклеотидов комплементарны дистальному концу разрыва РАМ-содержащей цепи и 91 нуклеотид – проксимальному концу[46]. Также по данным Richardson и соавторов использование донорной последовательности, комплементарный РАМ содержащей цепи, в 2,6 раза увеличивает эффективность HDR, по сравнению с использованием ssODN, комплементарного целевой цепи ДНК [49].
В отличие от ssODN, двуцепочечный плазмидный донор позволяет встраивать более крупные фрагменты ДНК. Линеаризация донора – типичный способ увеличения эффективности его интеграции в геном. Более того, на эффективность HDR можно влиять, используя разные способы линеаризации плазмиды. Так же, как в случае с ssODN, наиболее эффективна для гомологичной рекомбинации ассиметричная конструкция, у которой более короткое плечо с дистальной стороны от PAM. Таким способом удалось повысить эффективность нокина в человеческих индуцированных плюрипотентных стволовых клетках (чИПСК) в 4,2 раза.Еще один вариант линеаризации донора – двойное разрезание плазмиды, содержащей донорную последовательность. Подход заключается в следующем: с внешней стороны плеч гомологии вводят два сайта узнавания для той же sgRNA, которая будет направлять Cas9 к целевому сайту в геноме. После сборки sgRNA/Cas9 комплексов они «скринируют» как геномную, так и плазмидную ДНК в поисках сайтов узнавания для sgRNA и, находя, условно одновременно вносят DSB в геном и линеаризуют донор. Таким образом, происходит синхронизация потребности в матрице для гомологичной рекомбинации и ее доступности. При использовании описанной стратегии J.P. Zhang с коллегами (2017) удалось увеличить эффективность HDR в некоторых локусах чИПСК в 7.6 раза, а дополнительная синхронизация клеточного циклапривела к увеличению эффективности встройки еще в 1,5–2 раза. Важным преимуществом использования донора с двумя сайтами разрезания для sgRNA является возможность ограничиться короткими плечами гомологии (всего 600 п. о.) без потери эффективности нокина[50]. Однако линеаризация донора увеличивает, в том числе, и неспецифическую его интеграцию в геном[20].
Введение генетической конструкции в выбранный локус будет проходить с тем большей вероятностью, чем эффективнее происходит внесение DSB системой CRISPR/Cas9. Составные части системы CRISPR/Cas9 – sgRNA и Cas9 нуклеаза могут быть доставлены в клетку в разном виде, а именно: как плазмидная ДНК, sgRNA и Cas9 mRNA или рибонуклеиновый комплекс (RNP комплекс), состоящий из sgRNA и белка Cas9. Для легко трансфицирующихся клеток, таких как HEK293 или мышиные ЭСК, зависимость эффективности работы системы CRISPR/Cas9 от используемого вида ее составляющих минимальна. Для чИПСК же, наоборот, RNP комплекс в 4,3 и 2,7 раза позволяет увеличить эффективность внесения DSB по сравнению с использованием плазмид и sgRNA + Cas9 mRNA соответственно [51]. Наблюдаемое для некоторых линий клеток повышение эффективности нокина при использовании RNP комплекса также может быть связано с тем, что он активен сразу же после попадания в клетку. Быстрый запуск работы нуклеазы особенно принципиален при использовании линеаризованных доноров для HDR. Их концентрация в клетке наивысшая непосредственно после трансфекции, и с течением времени они достаточно быстро деградируют. Дополнительным преимуществом использования RNP комплекса является, в первую очередь, снижение нецелевых эффектов за счет быстрой деградации белка Cas9, которая происходит в течение 24 ч после трансфекции, во-вторых, снижение вероятности нецелевых встроек в геном, связанное с отсутствием ДНК вектора, несущего последовательности sgRNA и Cas9. Кроме того, трансфекция RNP комплекса для некоторых видов клеток менее токсична, чем плазмидная [52].
В 2015 году на фибробластах свиньи была продемонстрирована возможность использования вспомогательных матричных одноцепочечных олигонуклеотидов для запуска опосредованной гомологичной рекомбинация при ко-трансфекции с нуклеазным вектором[37]. Для редактирования были использованы фетальные фибробласты свиней породы ландрас и китайской породы эрхуалиан (Erhualian).
Векторы, кодирующие систему CRISPR/Cas9, были сконструированы на основе плазмиды Addgene(42230), состоящей из элементов экспрессии U6-sgRNA и Cas9с интеграцией двух различных направляющих последовательностей гомологичных третьему экзону гена MSTNсвиньи:
-sgRNA#1GАТАТAAGGССАAТТACTGCTCtgg;
-sgRNA#2 AАAGАTCCAGGAGAGATTTtgg.
Последовательность донорного олигонуклеотида: GGGTАTTTTTGTАAАААТАСAААТТСAСAСТСТССAGAGGССTCAGTAАTTGGCCTTАТАTCTTTTGGGTGCAAТАAТCCAGТСС (жирным шрифтом выделены добавочные нуклеотиды).
Фетальные фибробласты подвергали электропорации с использованием прибора BTX ECM 2001 в кюветах с зазором 2 мм (300 В, 1 мс, 3 импульса на 1 повтор). Приэтомиспользовалось 25 мкгплазмидной ДНК вектора CRISPR/Cas9 и 3 мкмоль донорного олигонуклеотида. Спустя 36 часов после электропорации Cas9, помещали в чашки Петри. Затем отдельные колонии клеток отбирали и культивировали в 24-луночных планшетах. После достижения конфлюентности 80–90 % колонии клеток субкультивировали и 10% каждой колонии подвергали индивидуальному лизированию. Лизат использовали в качестве матрицы для ПЦР. Используемыепраймеры:
прямой 5′-GGCGAAGACCTCAGGGAAATTTATATTG-3′;
обратный 5′-ACAGCGATCTACTACCATGGCTGGAATT-3
ПЦР проводилась по протоколу, приведенному в Таблице 1.
Таблица 1 – Протокол амплификации целевых фрагментов [37]
| Повторяемость | Этап | Температура, °C | Продолжительность, с |
| 1 цикл | Денатурация | 94 | 300 |
| 35 циклов | Денатурация | 94 | 15 |
| Отжиг | 62 | 10 | |
| Элонгация | 68 | 15 | |
| 1 цикл | Финальная элонгация | 72 | 480 |
Колонии клеток, признанные по результатам секвенирования мутантными, были размножены изаморожены в крио хранилище.
Спустя 24-часа после электропорации оценивали экспрессию pEGFP-N1 в фибробластах с использованием флуоресцентной микроскопии (Olympus BX51) с соответствующими фильтрами возбуждения. Собранные клетки дважды промывали, затем ресуспендировали в 300 мкл PBS и анализировали с помощью проточного цитометра BD Accuri C6.Эффективность редакции при использовании векторов по отдельности не превышала 21,79 %, при ко-трансфекции двух различных векторов эффективность составляла 31,5 %. Эффективность гомологичной рекомбинации для внесения ожидаемой инсерции составила только 3,64 %.Интересно, что частота одновременного расщепления двумя sgRNA оказалась выше ожидаемой. Возможно, что такая повышенная эффективность частично была связана с гомологически направленной репарацией, при которой один мутантный аллель служил матрицей для другого аллеля.
Из 51 колонии фибробластов, полученной от свиней породы ландрас и прошедшей процедуру трансфекции, 10 колоний несли интересующие мутации, из 37 колоний полученных от эрхуальских свиней – мутации несли 13 колоний. В качестве донорских клеток для SCNT были выбраны четыре колонии фибробластов предположительных нуль-мутантов по гену миостатина, полученных от свиней породы ландрас, несущие различные инсерции и делеции длиной от 1 до 29 п.о.
Процедуры SCNT и эмбриотрансфер выполнялись по стандартным протоколам. C использованием SCNT было получено 8 мутантных поросят-клонов с отредактированной последовательность третьего экзона гена MSTN: два мертворожденных и шесть умерших в течение первой недели после рождения.Живая масса при рождении у мутантных особей в среднем была на 15% больше, чем у однопометников с диким генотиипом. По результатам гистологических исследований у мутантов обнаружена гиперплазия мышечных волокон. Уровень экспрессии MSTN в сердечной мышце мутантных поросят был в разы ниже, чем в мышце поросят контрольной группы, в то время как уровень экспрессии MSTN в фибробластах значительно не различался [37].
В 2018 году опубликованы результаты эксперимента по направленному интегрированию последовательностей генов гуманизированного Fat-1 (humanized omega-3 fatty acid desaturase fat-1, hFat-1) и усиленного зеленого флуоресцентного протеина (enhanced green fluorescent proteine, GFP) в область гена миостатина КРС и гена β-казеина козы[30].Белок миостатин является негативным регулятором мышечного роста, а продукт гена hFat-1 регулирует выработку ω-3 полиненасыщенных жирных кислот (ПНЖК). Благодаря этомунокаут эндогенного миостатина при сайт-специфичном нокине с использованием последовательности гена hFat-1 может быть использован не только для увеличения мышечной массы сельскохозяйственных животных, но и для насыщения получаемого мяса ценными ПНЖК. Генβ-казеина (CSN2) активно экспрессируется в клетках молочной железы ивлияет на качественные характеристики получаемого молока. Внесение последовательностигенаhFat-1 вобластьгенаCSN2 может быть использовано для насыщения молока ПНЖК. НокингенаGFPприводиткнокаутуцелевыхгеновипозволяет судить об успехе геномного редактирования по наличию флуоресценции клеток в ультрафиолетовом свете. Для облегчения селекции клеток с отредактированным геномом, помимо последовательностей генов hFat-1 и GFP в клетки вносили последовательности гена устойчивости к неомицину.
Плазмида, кодирующаякомпоненты системы CRISPR/Cas9, направленная на разрезание экзона 2 гена MSTNКРС, была сконструирована с использованием последовательности гидовой РНКGGATTTTGAAGCTTTTGGATggg (строчные буквы обозначают PAM). Плазмида, кодирующаякомпоненты системы CRISPR/Cas9, направленная на разрезание экзона 2 гена CSN2 козы, была сконструирована с использованием гидовой последовательности CCTCATCCTTGCCTGTCTGGtgg (строчные буквы обозначают PAM).
На основе плазмиды GFP-PGK-NeoR было сконструировано четыре вида донорских плазмид:
– плазмида pbFlrkt с интегрированными последовательностями плечгомологии к гену MSTNКРС, фланкирующих ген hFat‑1 (длина 5’ плеча– 885 п.н., длина 3’ плеча – 823 п.н.);
– плазмида pbGlrkt с интегрированными последовательностями плеч гомологии к гену MSTN КРС, фланкирующих ген eGFP (длина 5’ плеча – 885 п.н., длина 3’ плеча – 823 п.н.);
– плазмида pgFlrkt с интегрированными последовательностями плеч гомологии к гену CSN2 козы, фланкирующих ген hFat‑1 (длина 5’ плеча – 1024 п.н., длина 3’ плеча – 1028 п.н.);
– плазмида pgGlrkt с интегрированными последовательностями плеч гомологии к гену CSN2 козы, фланкирующих ген eGFP (длина 5’ плеча – 1024 п.н., длина 3’ плеча – 1028 п.н.).
В качестве промотора использовался синтетический промотор CAG.
Сконструированные плазмиды были внесены в фибробласты коровы и козы с использованием двухимпульсной электропорации (200 В, 2 мс, кюветы 2мм, прибор NEPA21). Концентрацияиспользуемыхплазмидсоставляла 1 000 нг/мкл, соотношениевносимых донорных плазмид и плазмид, кодирующих системуCRISPR/Cas9 – 1:1. Длятрансфекции 9×105клеток, ресуспендированныхв 90 мклсреды Opti-MEMиспользовали 10 мкл плазмидной ДНК. Послеэлектропорацииклетки культивировали при температуре 38.5°Cи 5% концентрации CO2 в течение 48 часов. Затем для отбора успешно трансфицированных клеток в среду вносили препарат генетицин (аналог неомицина) и культивировали клетки еще 7 дней. За это время клетки образовали колонии с видимыми границами и появилась возможность выделения одноклеточных колонийдля дальнейшего анализа.
Для детекции нокина генов eGFPиhFat-1 использовалась полимеразная цепная реакция. Праймеры были разработаны таким образом, что позволяли судить об успешности гомологичной рекомбинации с использование донорных последовательностей.
Праймеры для детекции интеграции eGFP/hFat-1 в локус миостатина КРС:
–LH-LF-ACGGCTCCTTGGAAGACGATG;
–LH-LR-AATGGAAAGTCCCTATTGGCGTTA;
–LH-RF-CGATGCCTGCTTGCCGAATA;
– LH-RR-AGGAAGGTAGAGGGATGAAGATAGTGG
Праймеры для детекции интеграции eGFP/hFat-1 в локус β-казеина козы:
– LC-LF-TAATGGATTCTAGGTATTATGC
–LC-GFPLR-TGGTAATAGCGATGACTAATACG
–LC-GFPRF-CGATGCCTGCTTGCCGAATA
–LC-FATLR-AATGGAAAGTCCCTATTGGCGTTA
– LC-FATRF-ATCGCCTTCTATCGCCTTCTT
– LC-RR- GGAAACAACTGAAGTGACTTAGC
ПЦР для детекции интеграции генов eGFP/hFat-1 в целевые локусыпроводилась по протоколу представленному в Таблице 2.
Таблица 2– Протокол амплификации целевых локусов [30].
| Повторяемость | Этап | Температура, °C | Продолжительность, с |
| 35 циклов | Денатурация | 95 | 50 |
| Отжиг | 58/60 | 50 | |
| Элонгация | 72 | 120 | |
| 1 цикл | Финальная элонгация | 72 | 600 |
Прико-трансфекцииплазмидCRISPR/Cas9 иpbGlrktв фибробласты КРС было получено 57 клеточных линий с интегрированным геном eGFP. ЭффективностьCRISPR/Cas9-опосредованнойтрансфекцииeGFPсоставила 77,02 %.При ко-трансфекции плазмид CRISPR/Cas9 и pbFlrkt в фибробласты КРС было получено 64 клеточные линии с интегрированным геном hFat-1. ЭффективностьCRISPR/Cas9-опосредованнойтрансфекцииhFat-1составила 79,01 %.
При совместном использовании плазмиды CRISPR/Cas9 и донорной плазмиды pgFlrk было получено 26 клеточных линии с интеграцией последовательностиhFat-1 в область гена CSN2. При совместном использовании плазмиды CRISPR/Cas9 и донорной плазмиды pgGlrkt было получено 19 клеточных линии с интеграцией последовательностиeGFPв область гена CSN2. Эффективность интеграции hFat-1 и eGFP составила 74,29 % и 70,37 % соответственно.Результаты секвенирования генома модифицированных клеток в статье не приводятся[30].
В 2018 году опубликованы результаты работы по получению клонированных коз с одновременным нокаутом эндогенного MSTN и сайт-специфичном нокине экзогенного fat-1[34]. Для обеспечения биологической безопасности в эксперименте не использовались селективные гены флуоресценции или устойчивости к антибиотикам. На основе вектора Addgene (ID 41815) было сконструировано 2 вида плазмид, кодирующих единую химерную направляющую РНКс интеграцией различных гидовых последовательностей, гомологичных первому экзону гена миостатина козы(векторы sgRNA1-cgatgactaccacgttacga иsgRNA2-cgttacgacggaaacggtca).Донорный нокин-вектор fat-1 (5’h-pCAGDNA3-fat-1-3’h) содержал 5′-гомологичное плечо, промотор CAG, ген fat-1, последовательность поли A и 3 гомологичное плечо. Оба гомологичных плеча были сконструированы для целевых локусов sgRNA с длиной 1019 п.н. и 1023 п.н. соответственно.ПолученныеCRISR/Cas9 векторы путем электропорации трансфицировали в фетальные фибробласты козы одновременно с донорной плазмидой, несущей генfat-1(225 В в течение 2,5 мс). Длятрансфекциииспользовалось 4 мкглинеаризированногонокинвектораfat-1, 3 мкгплазмиды CRISPR/Cas9 и 3 мкг линеаризированной плазмиды sgRNA2. После 48 ч инкубации клетки собирали для экстракции геномной ДНК, полученную ДНК амплифицировали с помощью ПЦР. Последовательности используемых праймеров:
— MSТN-FCTATTTATGCTGCTTGTTGC;
— MSТN-R СТАТСТСССААТССТТСАСС;
— fat-1-F TTTCGCA CGGTTCCTCTG;
— fat-1-R СТТAGCAATTTCTTGCCTTT;
— 5′- junction-F Т ACCAGCACAGTAGTGAGAAGC;
— 5′ — junction-R GGGCTАTGAACTААTGACCCCG;
— 3 ‘ — junction-F TTTCGCA CGGTTCCTCTG;
— 3 ‘ — junction-R СТТ AGCAA TTTCTTGCCTTT.
После амплификации производили очистку ПЦР-продукта и оценивалиэффективность векторов sgRNA1 и sgRNA2 с помощью нуклеазного анализа Surveyor. Эффективность редактирования составила 11 % и 35 % соответственно. Таким образом, вектор sgRNA2 оказался более эффективным и был использован в последующих экспериментах.
Линии фибробластов, успешно прошедшие трансфекцию, по результатам секвенирования целевой области и типа выявленных модификаций были подразделены на три группы:
-клетки с нокаутом гена MSTN;
— клетки снокином гена fat-1;
— клетки с одновременным сайт-специфичным нокином гена fat-1инокаутом гена MSTN;
Для определения типа редакции продукты ПЦР-реакции, полученные от 156 моноклональных клеточных линий, были отправлены на секвенирование. Инсерция последовательности гена fat-1выявлена у представителей 101 клеточной линии. Эффективность интеграции составил 67,3 %
Для идентификации клеток с одновременным нокаутом гена MSTNи интеграцией гена Fat-1 была проведена ПЦР-реакцияс ДНК фибробластов 101 клеточной линии с подтвержденной интеграцией гена fat-1(использовались праймеры 5’- и 3’- junction). Последующеесеквенированиепоказало, что 40 клеточныхлинийимели одновременный нокаут гена MSTN и сайт-специфичный нокин гена fat-1, но все полученные модификации были моно аллельными. Эффективность CRISPR/Cas9 опосредованного генного нокаута и направленной интеграции гена fat-1 в область первого экзона гена MSTN составила 25,6 %.
Клеточная линия P011 была использована в качестве ядерного донора для получения мутантных коз при соматическом клонировании. Процедуры SCNT и эмбриотрансфер выполнялись по стандартным протоколам.CиспользованиемSCNTбылореконструировано 249 эмбрионов, 134 из них были трансфицированы 56 реципиентам. Беременность наступила у 8 животных, в результате был получен 1 трансгенный козленок. Его живая масса при рождении составила 2,6 кг, живая масса при отъеме 21,5 кг.
Наличие генных модификаций у полученных животных оценивалось с использованием ПЦР реакций с праймерами трех типов: к экзону гена MSTN, к плечам гомологии, к гену fat-1.Для оценки уровней РНК MSTN и fat-1 проводили ПЦР в реальном времени, в качестве внутреннего стандарта использовали РНК гена GAPDH.Условия проведения реакции подобраны с учетом инструкции, прилагаемой к набору TaKaRa REAL-TIME PCR.Используемые праймеры:
qPCR-MSТN-F TGTAACCTTCCCAGAACCAG;
qPCR-MSТN-R GCAATAATCCAATCCCATCC;
qPCR-fat-1-F GTCAAGTCCATCCGCTATCTTGT;
qPCR-fat-2-R CCGAACAGCCCGAAATACTC;
qPCR-GAPDH-F CGTGTCCGTTGTGGATCTGA;
qPCR-GAPDH-R GAGTGTCGCTGTTGAAGTCG.
Уставлено, что уровень экспрессии гена fat-1 у трансгенной козы был в 1,50 раза выше, чем у животного дикого генотипа, в то время как уровеньэкспрессии MSTNбыл в два раза ниже.
Определялось относительное содержание различных полиненасыщенных жирных кислот в фибробластах мутантного клона и животного с диким генотипом. Исследование выполнялось с использованием газовой хроматографии на GCMS-QP2010 Ultra (Shimadzu), оборудованном колонкой HP-88 (толщина пленки 0,20 мкмоль, 100,0 м × 0,25 мм); гелий использовали в качестве газа-носителя. Содержание ПНЖК n-6 в клетках мутантного животного было значительно ниже, чем в клетках животных контрольной группы, содержание кислот n-3 достоверно не различалось [34].
В 2020 году опубликованы результаты экспериментов по получению трансгенных эмбрионов КРС с одновременным нокаутом гена MSTNи сайт-специфичном нокине гена PPARγ(peroxisome proliferator‐activated receptor‐γ, γ-рецептор, активируемый пероксисомным пролифератором)[53]. Ген PPARγявляется положительным регулятором адипогенеза и активно экспрессируется в жировой ткани, регулирует экспрессию FABP4, FAS, C/EBPα, ACS и LPL. В отсутствии экспрессии гена PPARγ инициация адипогенной дифференцировки невозможна. Оверэкспрессия PPARγ может способствовать производству зрелых адипоцитов и заставлять миоциты/миоблаты дифференцироваться в адипоциты, тем самым улучшая отложение внутримышечного жира.Целью исследований являлось увеличение уровня экспрессии гена PPARγ при одновременном снижении уровня экспрессии гена MSTN для увеличения мышечной массы и отложения внутримышечного жира, и улучшения вкусовых характеристик получаемого мяса. Вкачествецелевойобластивыбранучасток в гене MSTN длиной 650 п.о. (chr2:6219940‐6220590). ВекторыPSpCas9(BB)‐2A‐GFP (PX458, Addgene)иT‐VectorpMD19 (Simple; Takara, Japan)былииспользованывкачествеосновыприконструированииCas9 расщепляющей плазмиды и донорной плазмиды для гомологичной рекомбинации соответственно.Использовалось несколько вариантов последовательностей направляющей РНК:
— sg 11 –TTGACCAGAACCAATTTAACAGG;
— sg 19 – TTGCAGTTGGCAAGGGTATATGG;
— sg 27 – GTCAGTCTCATTAAGTGCTCGGG;
— sg 29 – CTGACACACTTATCTGAGTTTGG;
— sg 30 – TGACACACTTATCTGAGTTTGGG;
— sg 32 – ACACACTTATCTGAGTTTGGGGG;
Сиспользованиемразработанныхконструкций в область гена миостатина была внесена мутация g + 6,354 G‐A,способствующая развитию мышечной гипертрофии путем микроРНК-опосредованного ингибирования трансляции гена при создании таргетного сайта для микроРНК mir1 и mir206. В ходе эксперимента в 3’‑фланкирующую область гена миостатина интегрировали последовательность трансгена CMV-PPARγ[53].
Проанализированные данные говорят о больших перспективах направленной интеграции заданных последовательностей в геном сельскохозяйственных животных с использованием системы CRISPR/Cas9 и донорных ДНК-матриц. Однако на данный моментэффективностьгомологичной рекомбинации остается очень низкой,дляполученияклонасжелаемойгенетическоймодификациейтребуетсяскрининг большого количества клеток (106–108).
3. Подготовка материально-технической базы для проведения экспериментов по редактированию генома
Эксперимент был выполнен на овцах северо-кавказкой мясо-шерстной породы в возрасте 1,5-2 года, со средней массой 48-54 кг. Все животные были клинически здоровыми и содержались в одинаковых условиях вивария факультета ветеринарной медицины и технологического менеджмента Ставрополького ГАУ.
Для проведения научно-исследовательской работы были подготовленысоответствующее реактивы (Таблица 3), расходные материалы и лабораторный инвентарь (Таблица 4).
Таблица 3 – Перечень реактивов используемых в лабораторных исследованиях
| № П/П | Наименование реактивов |
|---|---|
| DreamTaq™ Hot Start PCR Master Mix | |
| Dynabeads™ CD34 Набор для позитивной изоляции | |
| Dynabeads™ CD4 Набор для позитивной изоляции | |
| DynaMag™-5 Magnet | |
| GeneArt Набор для детекции геномного расщепления | |
| GeneRuler Express DNA Ladder | |
| GenPak® PCR Core (0,5мл) | |
| GlutaMAX добавки для клеточных культур | |
| O’RangeRuler 20 bp DNA Ladder | |
| Remel™ Минеральное масло | |
| TrackIt™ 1 Kb Plus DNA Ladder | |
| TrackIt™ 100 bp DNA Ladder | |
| TrackIt™ Cyan/Orange загрузочный буфер | |
| UltraPure™ TBE Buffer | |
| ZipRuler Express DNA Ladder Set | |
| Аминокислоты заменимые МЕМ | |
| Аминокислоты незам. МЕМ | |
| Амфотерицин В (2,5 мкг/мл) | |
| Антибактериального препарат Пенициллин-стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина) | |
| Вектор нуклеазы GeneArt ™ CRISPR с набором для обогащения CD4 (с компетентными клетками) | |
| Вода, свободная от нуклеаз | |
| ДНК-маркер O’RangeRuler™ 20 п.н., в растворе для нанесения на гель | |
| ДНК-маркер экспресс ZipRuler™ для нанесения на гель | |
| Добавка B-27™ Plus Supplement (50X) | |
| Желатин (кожа быка), тип.В, | |
| Изотонический разбавитель Diluent | |
| Коллагеназа 100 мг (тип 1) | |
| Коллагеназа 100 мг (тип 2 ) | |
| Липофектамин 3000 реагент для трансфекции, Lipofectamine 3000 | |
| Мастер-микс для ПЦР с ДНК-полимеразой DreamTaq Hot Start, с «горячим» стартом | |
| Набор GeneArt® CRISPR Nuclease Vector with OFP Reporter Kit (with competent cells), |
|
| Набор Plasmid Midiprep 2.0 | |
| Натрия пируват | |
| Питательная среда ДМЕМ жидкая, с глутамином и глюкозой 4,5г/л |
|
| Раствор Версена 0,45л | |
| Р-ор трипсин-ЭДТА | |
| Сыворотка эмбриональная телячья | |
| ТВЕ-буфер (Трис-борат-ЭДТА), UltraPureTM, | |
| Трипановый синий, 0,4 % раствор | |
| Трипсина раствор 0,25%, стер., 400 мл, | |
| Фосфатный буфер, рН 6,8 | |
| Фунгизон противогрибковый, жидкость | |
| Этидиум бромид, 10 мг | |
| Раствор этилового спирта 70 % и 96 % |
Таблица 4 – Перечень лабораторного инвентаряиспользуемого в лабораторных исследованиях
| № П/П | Наименование лабораторного инвентаря |
| Камера Горяева с покровными стеклами | |
| Конические стерильные полипропиленовые центрифужные пробирки, 50 мл | |
| Криогенные пробирки общего назначения для длительного хранения Nalgene, 15 мл | |
| Криогенные пробирки общего назначения для длительного хранения Nalgene, 5 мл | |
| Микроцентрифужные пробирки градуированные объемом 1,5 мл | |
| Микроцентрифужные пробирки градуированные объемом 1,5 мл | |
| Микроцентрифужные пробирки градуированные объемом 2,0 мл | |
| Наконечники до 50 мкл ,с фильтром | |
| Наконечники универсальные для дозаторов объемом 0,1-10 мкл | |
| Наконечники универсальные для дозаторов объемом 1000 мкл | |
| Наконечники универсальные для дозаторов с фильтром объемом 1000 мкл | |
| Наконечники универсальные для дозаторов с фильтром объемом 1000 мкл | |
| Наконечники универсальные для дозаторов с фильтром объемом 50 мкл | |
| Ножницы медицинские (глазные) с прямыми изакругленными браншами | |
| Пинцеты анатомические | |
| Планшеты 24-луночные культуральные | |
| Пробирки 15 мл, центрифужные, конические, с завинчивающейся крышкой, стерильные | |
| Пробирки 50 мл, центрифужные, конические, с завинчивающейся крышкой, стерильные | |
| Стерильный сосуд для слива | |
| Чашки Петри культуральные, d 60 мм |
Для проведения научно-исследовательской работы было подготовлено необходимое оборудование.
Ламинарный шкаф LABGARD NU-425-400E II класса биологической безопасности для защиты оператора, продукта и окружающей среды, с рециркуляцией воздуха, производства компании «NuAire», США.
Высокоскоростная центрифуга «СМ-50» для пробирок Eppendorf производитель компания ELMI (Латвия) использовалась для осаждения проб в малых объемах.
Твердотельный термостат «Bio TDB-100» с функцией охлаждения и нагрева производитель компания BioSan (Латвия) использовали для поддержания требуемой температуры реактивов и сред, а также жизнеспособности при выполнении работ в ламинарном боксе.
С помощью весов аналитических GR-202 (Япония) производили высокоточное взвешивание реактивов используемых в ходе лабораторных исследований.
Для выращивания и исследования клеточных культур применяли лабораторный инкубатор с атмосферой СО2.
Для работы с компетентными клетками использовался Шейкер‑инкубатор ES-20/60, производитель компания BioSan. Прибор обеспечивал плавное или интенсивное перемешивание проб в колбах, установленных на платформе.
Для учета результатов исследований по культивированию фибробластов использовали инвертированный микроскоп Olympus IX71.
Для наблюдения за трансфицированными клетками использовали микроскоп Olympus BX41, дополненный блоком флуоресценции.
Для выделения плазмидной ДНК (CRISPR3, CRISPR38) набором Plasmid Midiprep 2.0, Евроген использовали настольную высокоскоростную лабораторную центрифугу SIGMA 3K30 (Sigma Laborzentrifugenс, Германия), с ускорением свыше 60 000 g.
Мешалка VortexZX3 (VELP, Италия), предназначена для перемешивания различных веществ в пробирках с помощью встряхиваний и круговых движений (Рисунок 17). Вортекс ZX3 используется для длительного непрерывного перемешивания и для перемешивания с меняющейся частотой вибрации. С помощью вортекса перемешивали суспензию, включающую ДНК, растворы (нейтрализующий, лизирующий, промывочный, элюирующий, раствор для удаления эндотоксинов).
Выделение ДНК из биологического материала (кровь, культура клеток) проводилось в специальном ламинарном боксе производитель компания LAMSYSTEMS.
Для определения концентрации ДНК использовали прибор Nanophotometer «ImpLEN», обеспечивающий однородность образца с наивысшей степенью точности с наименьшим объемом образца всего 0,3 мкл. Полные возможности сканирования варьируются от 200 — 900 нм для быстрого и полного анализа образцов за 3,5 сек.
Для приготовления ПЦР смеси использовали ПЦР-бокс «Cleaner ПЦР UVC/T-M-AR» производитель компания BioSan.
Для амплификации целевых участков в гене MSTN использовали амплификатор Терцик «ДНК-технология».
Также применялся горизонтальный форез нуклеиновых кислот в агарозном геле с помощью источника питания PowerPac Basic «BIO-RAD».
Результаты гельэлектрофореза фрагментов ДНК определяли с помощью системы гель-документирования ChemiDoc XRS+ производитель компания Bio-Rad.
4. Компьютерный анализ и конструирование гидовых последовательностей РНК и матричных последовательностей трансгенов
Выбор целевой последовательности и последующие конструирование одноцепочечных олигонуклеотидов имеет критически важное значение и во многом определяет успешность эксперимента. Последовательностьмишенидолжна иметь длину от 19 до 20 нуклеотидов и прилегать к мотивуPAM (NGG) на 3 ‘конце. Необходимо убедится, что целевая последовательность не содержит участков, гомологичных с другими генами, так как это может увеличить нецелевые эффекты. По опубликованных данным, комплекс gRNA-Cas9 потенциально может сработать при несоответствии в 1–3 нуклеотида. Можно выбрать целевую последовательность, кодирующую смысловую последовательность локус-мишень или антисмысловую последовательность. Таким образом, можно генерировать CRISPR РНК в двух возможные ориентации при условии, что они соответствует требованиям PAM на 3’ концах[54].
В качестве целевого гена для редактирования с использованием системы CRISPR/Cas9 выбран ген миостатина, кодирующий один из ключевых регуляторов мышечного роста и развития. При блокировании действия миостатина наблюдается увеличение мышечной массы и повышение силовых характеристик скелетных мышц[55–57]. У овец ген миостатина расположен на второй хромосоме, имеет 3 кодирующих экзона и два интрона. Общая длина гена 2894 пар оснований. Длина белкового продукта MSTN после сплайсинга составляет 375 аминокислот (Рисунок 22). В С-концевой области (281-375 амк.) расположен функционально важный специфичный домен, определяющий принадлежность гена MSTN к семейству ростовых факторов Transforming growth factor β-family (TGF-β). В N-концевой части аминокислоты с 39 по 249 определяют последовательность гомодимера пропептида MSTN, который в дальнейшем взаимодействует с TGF-бета-связывающими белками и обеспечивает функциональную активность гена[58].
Таблица 5– Перечень наиболее перспективных целевых последовательностей для редактирования гена MSTN овец методом нокина с использованием системы CRISPR/Cas9
| Ранг | Целевая последовательность | Позиция | Цепь | GC % | MM0 | MM1 | MM2 | MM3 | Эффективность |
| 1 | CTACCACGTTACGACGGAAACGG | 2:118144772 | + | 50 | 0 | 0 | 0 | 0 | 59.69 |
| 2 | TGACCGTTTCCGTCGTAACGTGG | 2:118144775 | — | 55 | 0 | 0 | 0 | 1 | 69.18 |
| 3 | CGATGACTACCACGTTACGACGG | 2:118144766 | + | 50 | 0 | 0 | 0 | 2 | 74.25 |
| 4 | GCATGGTAGTAGATCGCTGTGGG | 2:118149402 | + | 50 | 0 | 0 | 0 | 2 | 60.43 |
| 5 | TCAATCAGTTCCCGGAGTGGAGG | 2:118144695 | — | 55 | 0 | 0 | 0 | 2 | 57.35 |
| 6 | AATCCTCAGTAAGCTTCGCCTGG | 2:118144619 | + | 50 | 0 | 0 | 0 | 2 | 49.80 |
| 7 | CGTACTGATCAATCAGTTCCCGG | 2:118144703 | — | 45 | 0 | 0 | 0 | 2 | 50.15 |
| 8 | CAGAACTACTCACACTCCGTGGG | 2:118144806 | — | 50 | 0 | 0 | 0 | 3 | 69.10 |
| 9 | GTCTCAGATATATCCACAGTTGG | 2:118146737 | — | 40 | 0 | 0 | 0 | 3 | 52.32 |
| 10 | CTACTCACACTCCGTGGGCATGG | 2:118144801 | — | 60 | 0 | 0 | 0 | 3 | 53.78 |
| 11 | TACCTTGTACCGTCTTTCATGGG | 2:118146810 | — | 40 | 0 | 0 | 0 | 3 | 43.57 |
| 12 | GGCATGGTAGTAGATCGCTGTGG | 2:118149401 | + | 55 | 0 | 0 | 1 | 2 | 64.19 |
| 13 | TATAAGGCCAATTACTGCTCTGG | 2:118149215 | + | 40 | 0 | 0 | 1 | 2 | 44.67 |
| 14 | ATCTTTGTAGGAGTACAGCAAGG | 2:118149317 | — | 40 | 0 | 0 | 0 | 4 | 58.92 |
| 15 | AAGGGCCGGCTGAACCTTTGGGG | 2:118149298 | — | 60 | 0 | 0 | 0 | 4 | 49.99 |
| 16 | AAAGACGGTACAAGGTATACTGG | 2:118146816 | + | 40 | 0 | 0 | 0 | 4 | 43.67 |
| 17 | TCAATGCCTAAGTTGGATTCAGG | 2:118146930 | — | 40 | 0 | 0 | 0 | 4 | 39.42 |
| 18 | GCAAGGGCCGGCTGAACCTTTGG | 2:118149300 | — | 65 | 0 | 0 | 0 | 4 | 29.62 |
| 19 | CAAGGGCCGGCTGAACCTTTGGG | 2:118149299 | — | 60 | 0 | 0 | 0 | 4 | 26.00 |
| 20 | GCAGAACTACTCACACTCCGTGG | 2:118144807 | — | 55 | 0 | 0 | 1 | 3 | 65.37 |
| 38 | CTGTGTAATGCATGCTTGTGG (sgRNA222) | 2:118144554 | + | 44 | 0 | 1 | 5 | 99 | 65.63 |
Примечание: MM0, MM1, MM2 и MM3 – количество возможных нецелевых транскриптов (с 0, 1, 2 ,3 несовпадениями, соответственно), с которыми может связываться анализируемая гидовая РНК вне целевого гена; жирным шрифтом выделены целевые последовательности, используемые в дальнейшей работе.
В ходе проведения анализа последовательности кодирующих областей гена миостатина с использованием специализированного электронного ресурса https://chopchop.cbu.uib.no было определено 113 возможных вариантов20-ти нуклеотидной целевой последовательности с прилегающим NGG мотивом. Инструменты используемого ресурса позволили получить информацию о нуклеотидной последовательности предлагаемых целевых сайтов с ранжированием от наиболее желательной мишени (ранг1) до наименее желательной (ранг 113) с учетом вероятности нецелевых эффектов и предполагаемой эффективности. Информация о наиболее перспективных целевых сайтах приведен в Таблице 5. Для дальнейшего нокина из 113 предполагаемых мишеней были отобраны три целевых последовательности ранга 1-3, а также целевая последовательность sg222, успешно используемая нами на предыдущих этапах исследования при нокауте гена миостатина, имеющая согласно оценке ранг 38. Все целевые последовательности располагаются в области первого экзона гена MSTN.
Для направленного нокина коротких добавочных последовательностей в выбранные целевые области было сконструировано четыре различных варианта одноцепочечных матричных последовательностей (ssODN).Построение ssODN осуществлялось с использованием электронного ресурса Edit-R HDR Donor Designer – oligo, разработанного британской компанией Horizon Discovery. Донорные последовательности построены для каждой из выбранных мишеней с интеграцией добавочных стоп-кодонов (Таблица 6).
Таблица 6 – Последовательности донорных одноцепочечных олигонуклеотидов
| Донор | Последовательность | Длинна, н. |
| ssODN1 | TGGAAGACGATGACTACCACGTTACGACGGaatAAACGGTCATTACCATGCCCACGGAGTGTG | 63 |
| ssODN2 | CGGCTCCTTGGAAGACGATGACTACCACGTttagaaTACGACGGAAACGGTCATTACCATGCCCAC | 66 |
| ssODN3 | GCTCCTTGGAAGACGATGACTACCACGTTAggtaagCGACGGAAACGGTCATTACCATGCCCACGG | 66 |
| ssODN38 | GTGGAAAAAAAGGGGCTGTGTAATGCATGCtgaTTGTGGAGACAAAACAATAAATCCTCAAGA | 63 |
Примечание: жирным шрифтом отмечены добавочные нуклеотиды
Последовательности используемых нуклеотидных вставок должны обеспечить интеграцию добавочных стоп кодонов в кодирующие регионы гена MSTN и, как следствие, обеспечить преждевременное прекращение трансляции гена и появление функционально неактивного белка.
Согласно результатам компьютерного анализа использование донорной последовательности ssODN1 приведет к замещению 117 кодона, кодирующего аминокислоту треонин на стоп-кодон (Рисунок 1).

Рисунок 1.Изменение аминокислотной последовательности белка миостатина при интеграции добавочных нуклеотидов ssODN1
При успешной интеграции донорной последовательности ssODN2 114 кодон, кодирующий аминокислоту треонин заменится на стоп-кодон(Рисунок 2).

Рисунок 2.Изменение аминокислотной последовательности белка миостатина при интеграции добавочных нуклеотидов ssODN2
Использование донорной последовательности ssODN3 позволить заменить кодоны114 и 115, кодирующие аминокислоту треонин на кодон, кодирующий аминокислоту аргинин и стоп-кодон соответственно (Рисунок 3).

Рисунок 3.Изменение аминокислотной последовательности белка миостатина при интеграции добавочных нуклеотидов ssODN3
Донорная последовательность ssODN38разработана таким образом, что ееиспользование должно приводить к замене кодона 43, кодирующего аминокислоту лейцин на стоп-кодон. (Рисунок 4).

Рисунок 4.Изменение аминокислотной последовательности белка миостатина при интеграции добавочных нуклеотидов ssODN38
Для амплификации и последующего молекулярно-генетического анализа последовательностей-мишеней, отредактированных с использованием донорных ssODNподобраны соответствующие праймеры (Рисунок 5, Таблица 7).НаРисунке 5 графически представлено взаимное расположение целевых последовательностей и подобранных праймеров.
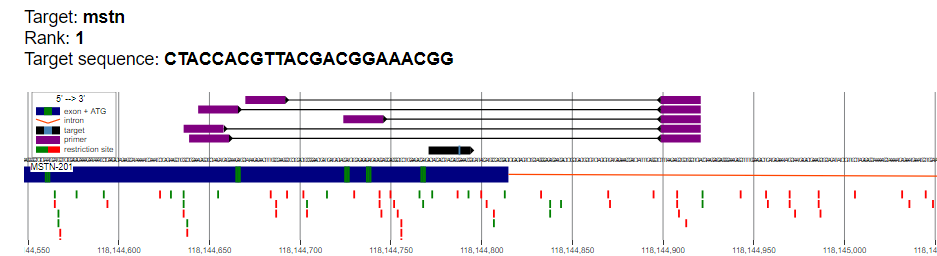
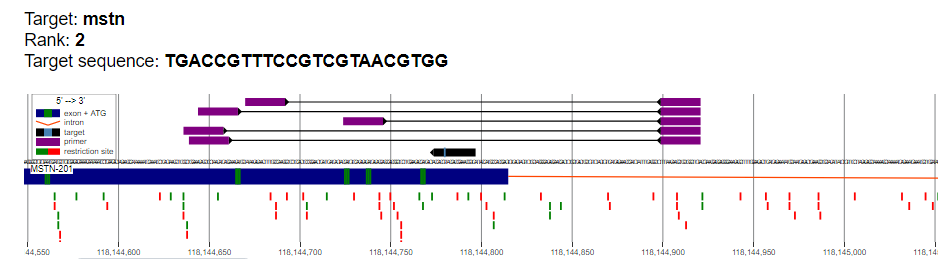
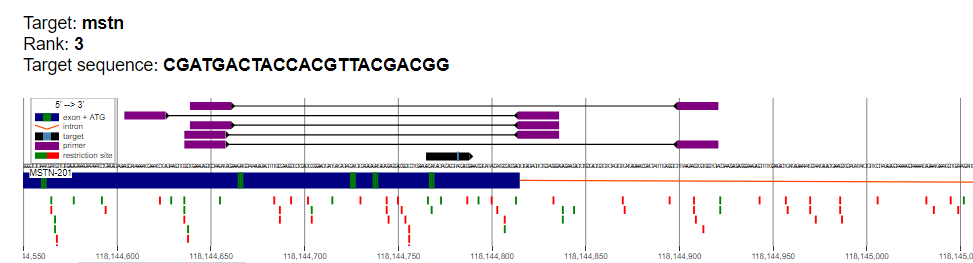
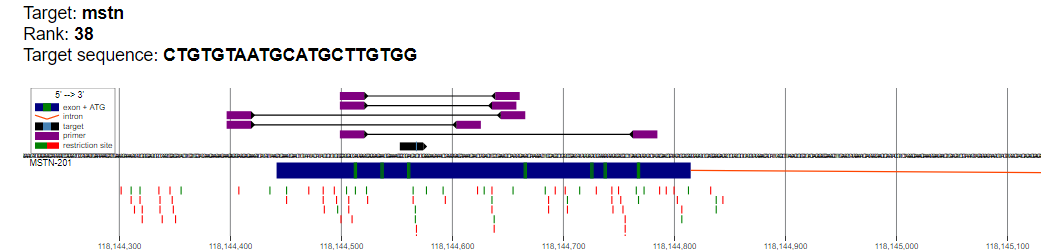
Рисунок 5. Графическое представление расположения отобранных целевых последовательностей и соответствующих праймеров
Таблица 7 – Праймеры для амплификации целевых областей
| Целевая последовательность 1 ранга (sgRNA1) | ||||||||
| Пара | Координаты FP | FP | FP Tm | Координаты RP | RP | RP Tm | Длина ампликона | |
| 1 | 2:118144637-118144659 | CCTGGAAACAGCTCCTAACATC | 60.1 | 2:118144900-118144922 | TACAAGCCAGCAGCTTGTTAAA | 60.1 | 285 | |
| 2 | 2:118144725-118144747 | GATGTCCAGAGAGATGACAGCA | 60.4 | 2:118144900-118144922 | TACAAGCCAGCAGCTTGTTAAA | 60.1 | 197 | |
| Целевая последовательность 2 ранга (sgRNA2) | ||||||||
| Пара | Координаты FP | FP | FP Tm | Координаты RP | RP | RP Tm | Длина ампликона | |
| 1 | 2:118144637-118144659 | CCTGGAAACAGCTCCTAACATC | 60.1 | 2:118144900-118144922 | TACAAGCCAGCAGCTTGTTAAA | 60.1 | 285 | |
| 2 | 2:118144725-118144747 | GATGTCCAGAGAGATGACAGCA | 60.4 | 2:118144900-118144922 | TACAAGCCAGCAGCTTGTTAAA | 60.1 | 197 | |
| Целевая последовательность 3 ранга (sgRNA3) | ||||||||
| Пара | Координаты FP | FP | FP Tm | Координаты RP | RP | RP Tm | Длина ампликона | |
| 1 | 2:118144637-118144659 | CCTGGAAACAGCTCCTAACATC | 60.1 | 2:118144900-118144922 | TACAAGCCAGCAGCTTGTTAAA | 60.1 | 285 | |
| 2 | 2:118144640-118144662 | GGAAACAGCTCCTAACATCAGC | 60.3 | 2:118144815-118144837 | TGCCCTAGCAGAACTACTCACA | 60.1 | 197 | |
| Целевая последовательность 38ранга (sgRNA222) | ||||||||
| Пара | Координаты FP | FP | FP Tm | Координаты RP | RP | RP Tm | Длина ампликона | |
| 1 | 2:118144500-118144522 | CCAGTGGATCTGAATGAGAACA | 60.1 | 2:118144764-118144786 | TCGTAACGTGGTAGTCATCGTC | 60.1 | 286 | |
| 2 | 2:118144398-118144420 | GGAAGCAGTAAGAGCAAGGAAA | 60.0 | 2:118144605-118144627 | TGAGGATTTGGATTTTTATGGC | 60.1 | 229 | |
Примечание: FP – прямойпраймер (forwardprimer), FR – обратныйпраймер (reversprimer)
Донорные плазмиды были сконструированы на основе вектора pTurboGFP-C, включающего в себя последовательность модифицированного зеленого флуоресцентного белка TurboGFP под цитомегаловирусным промотором PCMV IE. Длина 5’ гомологичного плеча составила 570 п.н., без учета адаптерной последовательности. Длина 3’ гомологичного плеча составила 971 п.н. без учета адаптерной последовательности. Для амплификации гомологичных фрагментов ДНК и их последующего клонирование в вектор были разработаны специфичные праймеры с адаптерными последовательностями (Таблица 4).
Таблица 8– Последовательности разработанных праймеров
| 5’ гомологичное плечо | ||
| Праймеры | адаптер | |
| прямой | TTCATTGTGGAGCAAGAGCCAA | TTCATTGTGGACATGT |
| обратный | GCTGGTGGCAGCGTGAGCAAGGGCGA | ACATGTGTTACGAC |
| 3’ гомологичное плечо | ||
| прямой | AAACGGTCATTACCATGCCCAC | AAACGGTCACGCGT |
| обратный | AGACAGGCACCTCAACAGAGAA | ACGCGTTCAACAGAGAA |
С использованием программного обеспечения Molecular Cloning Designer Simulator 1.022 был проведен компьютерный анализ последовательностей плеч гомологии и плазмиды, кодирующей ген eGFP. В ходе анализа были определены возможные варианты рестрикции последовательностей и их последующего лигирования (Рисунки 6,7).
Согласно данным компьютерного анализа, в процессе клонирования последовательности 5’ гомологичного плеча в вектор pTurboGFP-C, на этапе лигирования рестрикционных фрагментов возможно образование11 различных видов последовательностей кольцевой формы, диной от 22 до 9402 п.н. (MultiLig 22 –MultiLig 9402). При этом для дальнейшей работы должен быть использован векторMultiLig5276, имеющий длину 5276 п.н (Рисунок 6). В процессе клонирования 3’ гомологичного плеча в вектор MultiLig5276 на этапе лигирования также возможно образование 11 различных видов последовательностей кольцевой формы, при этом их длина составляет 28-1056 п.н. Для дальней работы должен быть использован вектор Muitiig 6248 (готовая донорная плазмида, Рисунок 8). Таким образом, при успешном последовательном клонировании плеч гомологии в вектор pTurboGFP-C, его длина увеличивается с 4701 до 6242 п.н.
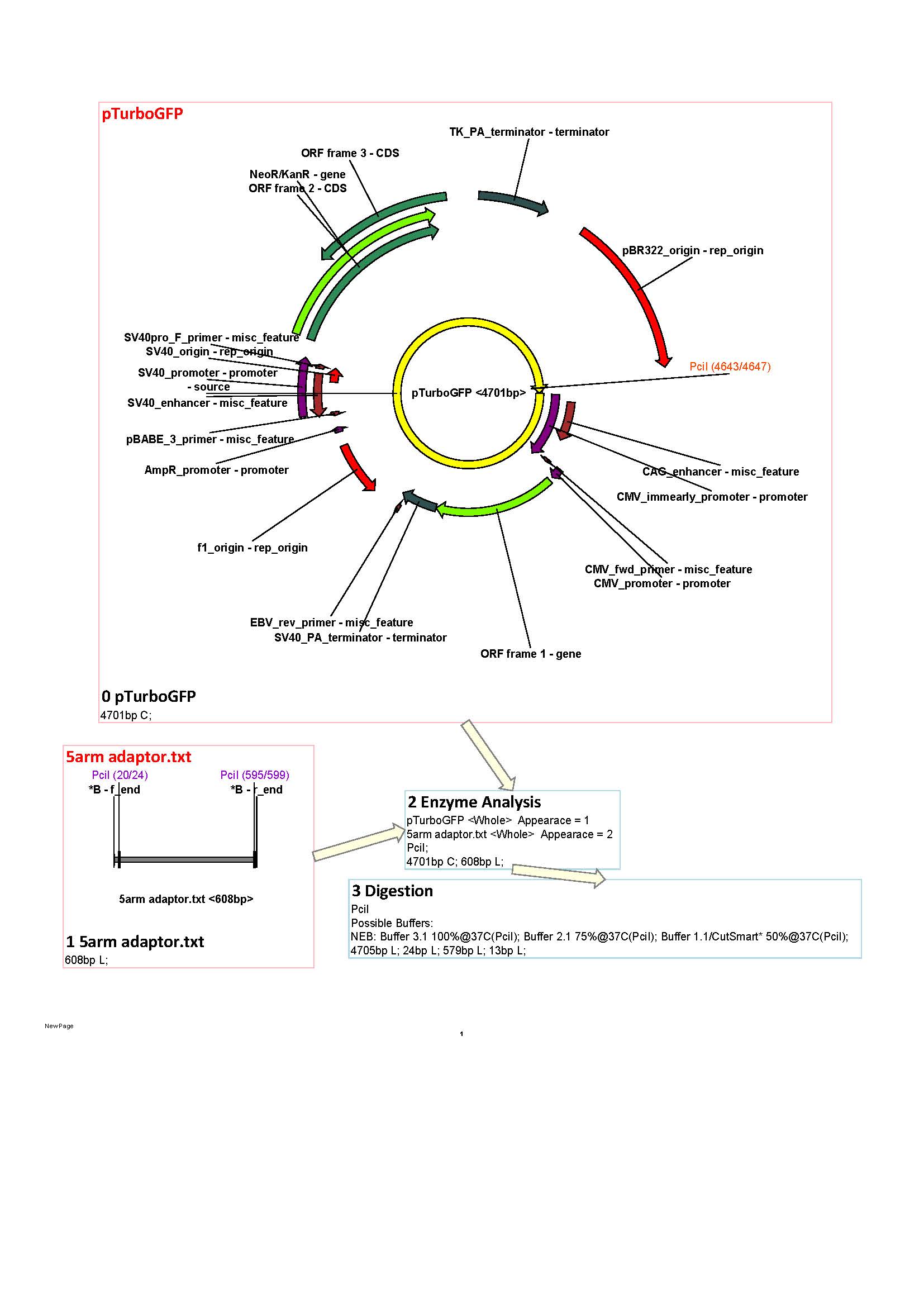
Рисунок 6.Общие сайты рестрикции для фермента PclIв последовательности 5’ плеча гомологии и вектора pTurboGFP-C
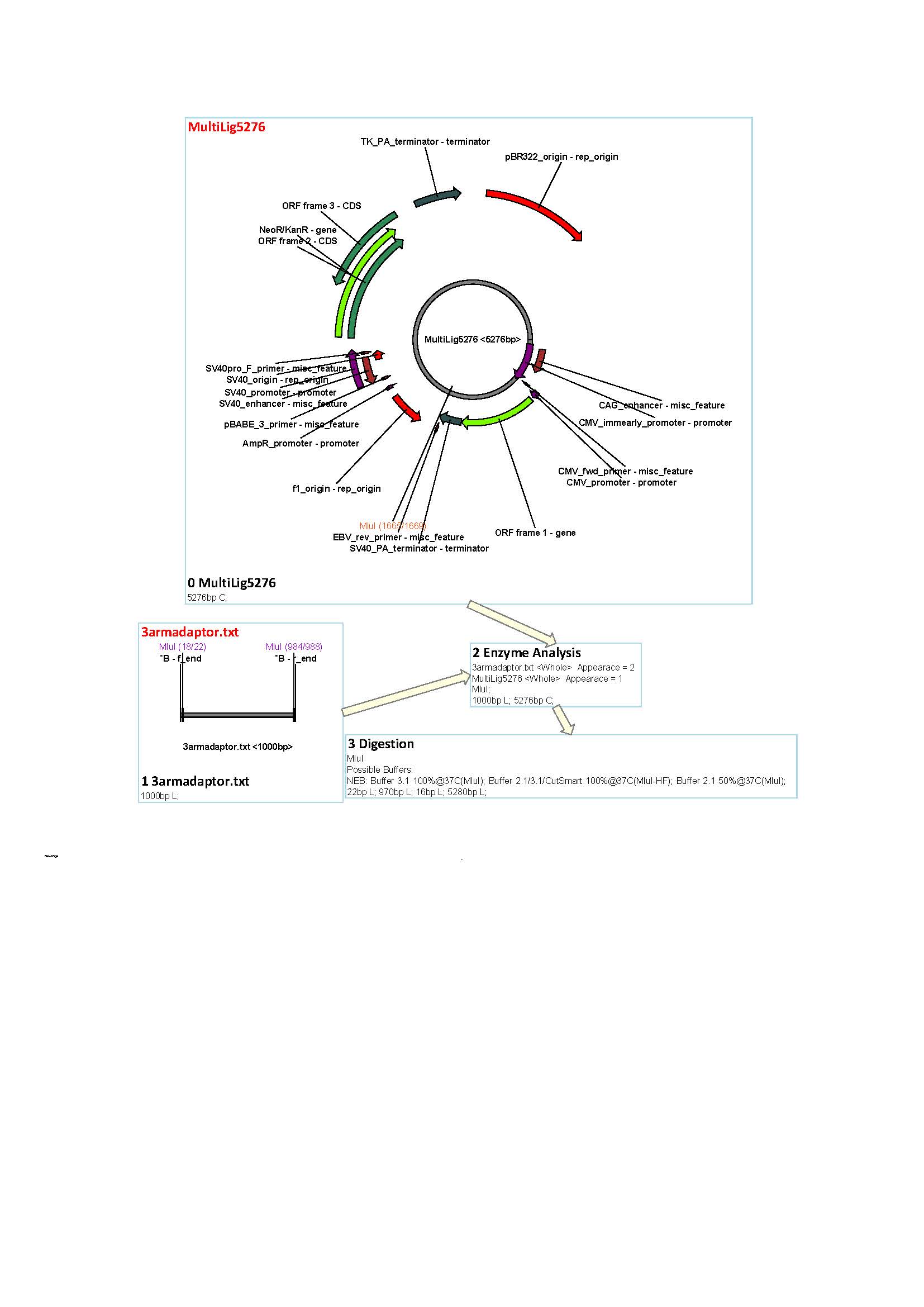
Рисунок 7. Общие сайты рестрикции для фермента МлuI в последовательности 3’ плеча гомологии и модифицированного вектора pTurboGFP-C (MultiLig5276)
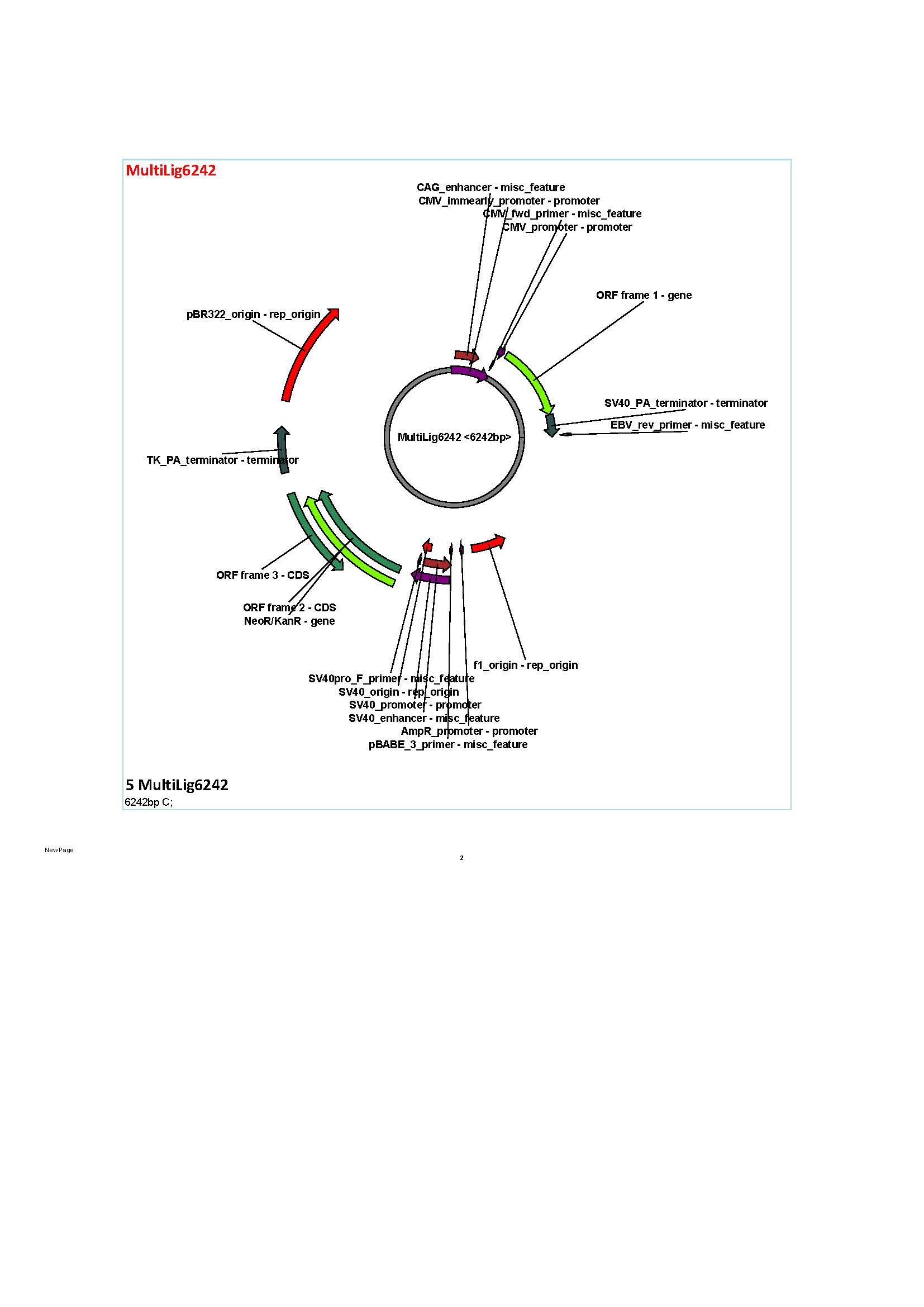
Рисунок 8.Графическое отображение донорной плазмиды MultiLig6242 (модифицированного вектора pTurboGFP-C)
В результате проведенного компьютерного анализа определено четыре различных варианта целевых последовательностей для редактирования с использованием системы CRSPR/Cas9. Сконструировано четыре варианта однонуклеотидных донорных последовательностей, несущих добавочные стоп-кодоны и один двуцепочечный донорный вектор, несущий последовательность гена вставки eGFP, фланикрованную плечами гомологии.
5. Получение олигонуклеотидов и плазмидных векторов, кодирующих систему CRISPR/CAS
Для редактирования гена миостатина у овец использовался коммерческий набор GeneArt® CRISPR Nuclease Vector Kit, включающий в себя реактивы, плазмидные векторы и компетентные клетки.
Для использования GeneArt® CRISPR Nuclease Vector Kit были разработаны четыре пары одноцепочечных олигонуклеотидов с хвостами, комплементарными участкам линеаризованного вектора; один, два кодирующих целевую РНК CRISPR (целевая последовательность, прямая цепь) и два других– их комплементы (обратная цепь). Затем был проведен отжиг олигонуклеотидов прямой и обратной цепей для получения двухцепочечного олигонуклеотида (ds oligonucleotide), подходящего для клонирования в линеаризованный вектор, входящий в набор.
После выбора целевой последовательности из 18 пар оснований было проведено конструирование специфичных для crRNA олигонуклеотидных праймеров (Таблица 9).
Таблица 9– Перечень синтезируемых последовательностей
| Наимено-
Вание |
Целевая последовательность (5’- 3’) | PAM | Цепь | Синтезируемая последовательности (5’- 3’) |
| sgRNA1.1 | CTACCACGTTACGACGGAAA | CGG | + | CTACCACGTTACGACGGAAAGTTTT |
| sgRNA1.2 | tttccgtcgtaacgtggtagcggtg | |||
| sgRNA2.1 | TGACCGTTTCCGTCGTAACG | TGG | — | TGACCGTTTCCGTCGTAACGGTTTT |
| sgRNA2.2 | cgttacgacggaaacggtcacggtg | |||
| sgRNA3.1 | CGATGACTACCACGTTACGA | CGG | + | CGATGACTACCACGTTACGAGTTTT |
| sgRNA3.2 | tcgtaacgtggtagtcatcgcggtg | |||
| sgRNA38.1 | CTGTGTAATGCATGCTTG | TGG | + | CTGTGTAATGCATGCTTGGTTTT |
| sgRNA38.2 | caagcatgcattacacagcggtg |
Для направленного клонирования ds-олигонуклеотида в нуклеазный вектор GeneArt® CRISPR к 3′-концам соответствующих одноцепочечных нуклеотидов было добавлено по 5 нуклеотидов. К 3′-концу олигонуклеотидов прямой цепи были добавлена последовательность GTTTT, комплементарная последовательности выступа, CAAAA, в линеаризованном нуклеазном векторе CRISPR и составляющая первые 5 оснований tracrRNA. Олигонуклеотид нижней цепи должен быть обратно комплементарен последовательности-мишени. К олигонуклеотидам обратной цепи, к 3′-концу были добавлены неуклеотиды CGGTG. Эта последовательность комплементарна последовательности выступа, CACCG, в линеаризованном нуклеазном векторе GeneArt® CRISPR и составляет последние 4 основания промотора U6 и первое основание, необходимое для стартового сайта транскрипции PolIII.
Далее был проведен отжиг двух одноцепочечных олигонуклеотидов для получения двухцепочечного олигонуклеотида с совместимыми концами для клонирования в нуклеазный вектор GeneArt® CRISPR.
Получение двухцепочечного олигонуклеотида
Необходимые материалы:
Прямая последовательность (200 мкмольв водном растворе или с буфером TE)
Обратная последовательность(200 мкмольв водном растворе или с буфером TE)
50 мкмольстокового раствора контрольных ds олигонуклеотидов
10XБуфер для отжига олигонуклеотидов
Вода, свободная от ДНКазы / РНКазы (входит в комплект)
1.5 млстерильные микроцентрифужные пробирки
95°Cтепловой блок
Процедура отжига
1. Вносили в чистую пустую пробирку при комнатной температуре следующие реагенты:
Прямая последовательность (200 мкмоль) 5 мкл
Обратная последовательность(200 мкмоль) 5 мкл
10XБуфер для отжига олигонуклеотидов 2 мкл
Вода, свободная от ДНКазы / РНКазы 8 мкл
Общий объем 20 мкл
2. Инкубировали пробирку при 95 ° С в течение 4 минут в термостате.
3. Доставали пробирку из термостата и давали реакционной смеси остыть до 25 ° C в течение 5–10 минут.
5. Центрифугировали пробирку в течение 5 секунд, осторожно перемешайте.
6. Для длительного хранения исходный раствор олигонуклеотида в концентрации 50 мкмоль замораживали при -20 ° C.
7. После отжига одноцепочечных олигонуклеотидов и олигонуклеотидов, контролирующих клонирование, выполняли два 100‑кратных серийных разведения исходного раствора олигонуклеотидов с концентрацией 50 мкмоль ds для приготовления исходного раствора олигонуклеотидов с концентрацией 500 нмоль ds (100-кратное разведение) и работали в дальнейшем с олигонуклеотидами ds в концентрации 5 нмоль. раствор (10 000-кратное разбавление).
Подготовка раствора dsОлигонуклеотидов 500 нмоль (стоковый раствор)
Подготовили раствор двухцепочечных нуклеотидов в концентрации 500 нмоль путем разбавления стокового раствора нуклеотидов (концентрация 50 мкмоль) в 100 раз.
1. Смешивали в микроцентрифужной пробирке:
50 мкмольстоковый раствор ds олигонуклеотидов1 мкл
Вода, свободная от ДНКазы / РНКазы 99 мкл
Общийобъём 100 мкл
2. Тщательно перемешивали на вортексе. Раствор готов. Для длительного хранения 500 нмоль стоковый раствор двуцепочечных олигонуклеотидов замораживали при температуре –20°C.
Подготовка рабочего раствора ds Олигонуклеотидов 5 нмоль
Подготовливали раствор двухцепочечных нуклеотидов в концентрации 5 нмоль путем разбавления стокового раствора нуклеотидов (концентрация 500 нмоль) в 100 раз.
1. Смешивали в микроцентрифужной пробирке:
500 нмольds раствор ds олигонуклеотидов 1 мкл
10XБуфер для отжига олигонуклеотидов 10 мкл
Вода, свободная от ДНКазы / РНКазы 89 мкл
Общийобъём 100 мкл
Оттаивание замороженных растворов олигонуклеотидов проводили на льду поскольку нагревание растворов двухцепочечных олигонуклеотидов приведет к их частичной денатурации и снижению эффективности клонирования.
Лигирование
После получения растворов двухцепочечных олигонуклеотидов в нужной концентрации клонировали синтезированные последовательности в нуклеазный вектор GeneArt® CRISPR. Векторлинеаризованный, имеет общую длину 9220 пар нуклеотидов, содержит последовательности, кодирующие белок Cas9 и зеленый флуоресцентный белок, сигналы ядерной локализации и ряд промоторов, обеспечивающих экспрессию CRISPR/Cas9 (Рисунок 9).
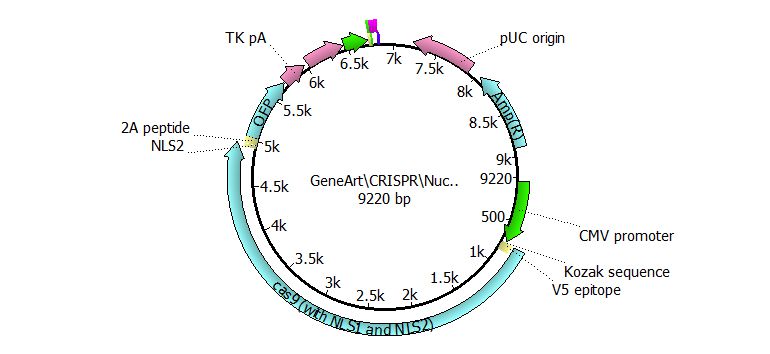
Рисунок 9. Графическое представление нуклеазного вектора GeneArt® CRISPR
Вектор сконструирован таким образом, что синтезированный двуцепочечный олигонуклеотид благодаря специфичным последовательностям на 3’-концах встраивается в позицию crRNA между последовательностями, кодирующими U6 промотор и трансактивирующую РНК (Рисунок 10).

Рисунок 10. Участок нуклеазного вектора GeneArt® CRISPR предназначенный для встраивания последовательности crRNA
После клонирования в вектор двухцепочечного олигонуклеотида, полученного отжигом последовательностей sgRNA1.1, sgRNA1.2и лигирования вектор замкнется в кольцо. Этот вид вектора получил название CRISPR 1 (по используемым олигонуклеотидам) (Рисунок 11).
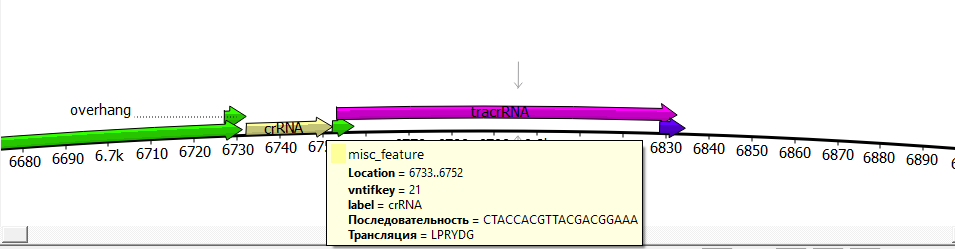
Рисунок 11. Участок нуклеазного вектора CRISPR 1, содержащий специфичную направляющую последовательность
В результате клонирования в вектор двухцепочечного олигонуклеотида, полученного отжигом последовательностей sgRNA2.1, sgRNA2.2 получена замкнутая кольцевая плазмида СRISPR 2 (Рисунок 11).

Рисунок 12. Участок нуклеазного вектора CRISPR 2, содержащий специфичную направляющую последовательность
В результате клонирования в плазмиду двухцепочечного олигонуклеотида, полученного отжигом последовательностей sgRNA3.1, sgRNA3.2 получен замкнутый кольцевой вектор СRISPR 3 (Рисунок 13).
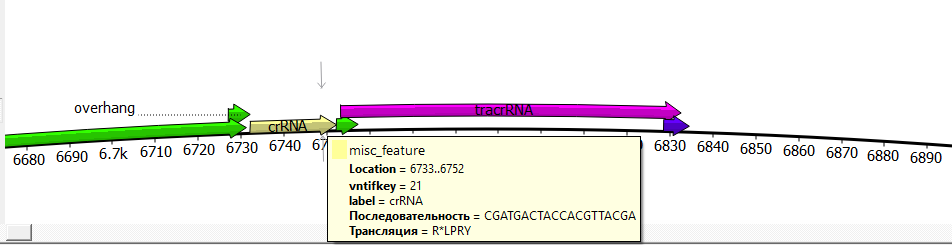
Рисунок 13. Участок нуклеазного вектора CRISPR 3, содержащий специфичную направляющую последовательность
После клонирования в вектор двухцепочечного олигонуклеотида, полученного отжигом последовательностей sgRNA38.1, sgRNA38.2 и лигирования вектор замкнется в кольцо. Этот вид вектора получил название CRISPR 38 (CRISPR222) (Рисунок 14).
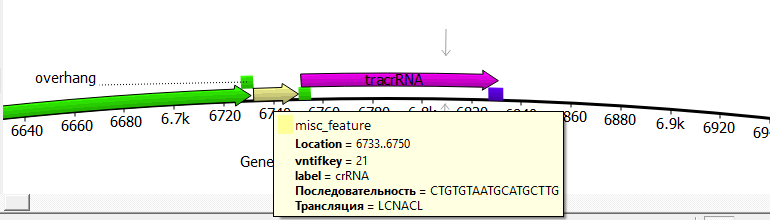
Рисунок 14. Участок нуклеазного вектора CRISPR 38 (CRISPR222), содержащий специфичную направляющую последовательность
Необходимые материалы
Раствор двухцепочечных олигонуклеотидов (5 нмольв 1XБуфер для отжига олигонуклеотидов; оттаивание проводить на льду)
Раствор контрольных двухцепочечных олигонуклеотидов (5 нмольв 1XБуфер для отжига олигонуклеотидов; оттаивание проводить на льду)
Линеаризированный вектор GeneArt® CRISPR Nuclease Vector
5XБуфер для лигирования(входит в комплект)
Вода, свободная от ДНКазы / РНКазы (входит в комплект)
T4 ДНК лигаза (входит в комплект)
Контроль:
В качестве положительного контроля в эксперименте по лигированию использовался ds олигонуклеотидпоставляемый с набором.
Контрольный олигонуклеотид ds поставляется в виде исходного стокового раствора 50 мкмольв 1X буфере для отжига олигонуклеотидов, и его необходимо повторно отжечь и разбавить в 10000 раз перед использованием в реакции лигирования.
Процедура лигирования
Общий объем смеси для лигирования каждого клонированного ds oligonucleoti составляет 20 мкл.
Реакция лигирования проходит при комнатной температуре.
1. Внесили в чистую микроцентрифужную пробирку компоненты реакции в следующем порядке:
5X Буфер для лигирования 4 мкл
Линеаризированный вектор GeneArt® CRISPR Nuclease Vector 2 мкл
ds oligonucleotide (5 нмоль) 2 мкл
Вода, свободная от ДНКазы / РНКазы 11 мкл
T4 ДНК лигаза1 мкл
Общий объём реакции 20 мкл
2. Тщательно перемешивали реакционную смесь пипетированием, не используя вортекс.
3. Инкубировали смесь в течение 10 минут при комнатной температуре (25–27 ° C).
Примечание: время инкубации может быть увеличено до 2 часов для увеличения выхода реакции.
4. Затем реакционную пробирку помещали на лед и переходили к трансформации компетентных клеток Transform One Shot ® TOP10 Competent E. Coli.
6. Клонирование генных конструкций в компетентных клетках
Для наработки достаточного для экспериментов с эукариотическими клетками количества собранных нуклеазных векторов CRISPR1, CRISPR2, CRISPR3, CRISPR38 выполнялось их клонирование в компетентных клетках E. Coli One ShotR, поставляемых в составе набора GeneArt® CRISPR Nuclease Vector Kit.
Для клонирования генных конструкций использовали следующие расходный материалы,стерильную посудуи оборудование:
1. Емкость со льдом
2. Чашки Петри (диаметром 5 см) одноразовые
3. Пробирки бактериологические (объемом от 10 до 20 мл)
4. Пипетки градуированные (пластиковые) (1, 2, 5, 10 мл)
5. Флаконы (пробирки) объемом 10 мл
6. Стерильный сосуд для слива
7. Шейкер-инкубатор
8. Термостат
9. Сосуд Дьюара
10. Твердотельный термостат
11. СО2 -инкубатор (термостат) с температурой 37О С
12. Центрифуга на 1000-2000 об /мин
13. Микроскоп инвертированный
14. Спектрофотометр
15. Ламинарный шкаф
16. Бактериальные петли
Также на данном этапе работы применяли растворы, питательные среды, сыворотки, антибиотики:
1. Компетентные клетки OneShotR
2. Плазмидная ДНК или продукты лигирования
3. Среда SOC
4. LBагар
5. LBбульон
6. Ампициллин
7. Стерильный глицерин
Трансформацию клеток выполняли по следующей схеме:
1. Поместили на лед пробирки с компетентными клетками до полного размораживания содержимого из расчета одна пробирка на трансформацию (50 мкл клеток One ShotR). Примерно 10 – 15 минут.
2. Добавили в каждую пробирку 3 мкл образца ДНК (плазмидная ДНК, продукты лигирования). Аккуратно перемешали содержимое легким встряхиванием.
3. Немедленно перенесли пробирки на лед. Инкубировали пробирки на льду в течение 30 минут.
4. Затем перенесли пробирки в твердотельный термостат (42 °С) и инкубировали в течение 30 секунд. Без встряхиваний.
5. Из твердотельного термостата пробирки (БЫСТРО!) перенесли на лед и инкубировали 3 минуты.
6. Добавили 250 мкл среды SOC (возможно использование LB бульона) комнатной температуры, перемешали содержимое.
7. Поместили пробирки в шейкер-инкубатор и инкубировали в течение 1 часа при 37 °С и 225 об/мин.
8. Высеяли 50 мкл реакционной смеси на предварительно нагретые чашки Петри с LB-агаром, содержащие 100 мкг/мл ампицилина. Оставшуюся часть реакции нанесли на вторую чашку Петри с LB-агаром (предварительно подогретую). Чашку Петри инкубировали в течение ночи при 37 °С. Поскольку векторGeneArt® CRISPR Nuclease Vector несет ген устойчивости к ампицилину, бактерии, успешно пережившие трансформацию и принявшие векторы, выжили и в дальнейшем образовали колонии. Остальные клетки погибли.
9. На следующие сутки отбирали 2-3 колонии (рисунок 31), помещали их в бульон и ставили в термостат на доращивание (14-18 часов). После чего выделяли плазмидную ДНК – чистый вектор для трансфекции эукариотических клеток. Перед выделение ДНК готовили библиотеку клеток (по протоколу криоконсервация библиотеки клеток).
10. Перед выделением ДНК проверяли бульон на мутность на спектрофотометре при длине волны 600 нм («мутность суспензии»).
Рисунок 15. Колонии бактерий в чашке Петри
Криоконсервацию библиотеки клеток осуществляли по следующему протоколу. В ламинарном шкафу бактериальной петлей набрали колонию бактерий и перенесли ее в пробирку с 2 мл LB бульона. Инкубировали в течение ночи (14–18 ч) при 37 °С в шейкер инкубаторе 250 об. мин. Пока культура не достигнет стационарной фазы (среда станет мутной).Смешали 0,85 мл культуры с 0,15 мл стерильного глицерина и перенесли в криопробирку. Перемешали в шейкер-инкубаторе. Концентрация глицерина может варьировать от 30 до 50 %.Далее пробирки перенесли в сосуд Дьюара и заморозили при -80 °С.
7. Выделение и очистка плазмидных векторов
Для выделения плазмидной ДНК из культуры клеток E. Coli применяли набор Plasmid Midiprep 2.0 производитель компания «Евроген». Выделение ДНК основано на применении микроцентрифужных колонок Midi с сорбционными стекловолокнистыми мембранами, предназначенных для центрифугирования. Основные свойства набора: емкость колонки составляет 500 мкг плазмидной ДНК; спектральный показатель препарата плазмидной ДНК A260/А280> 1,85; время выделения от 1 часа 30 мин.
Перед выделением ДНК бактериальную культуру инкубировали 16–20 часов в термостатируемом шейкере при 37°C, при постоянном перемешивании 200–250 об/мин. Следует избегать “перероста” бактериальной культуры, так как это может привести к большому количеству погибших клеток и к разрушению плазмид или загрязнению их хромосомными ДНК. плотность бактериальной культуры на спектрофотометре (OD600). Плотность бактериальной культуры контролировали с использованием спектрофотометра (OD600). По окончании роста на длине волны 600 нм показатель OD600 должен быть не менее 1,4‑1,6, плотность клеток 3–5 x 109 клеток/мл. Для плазмид с разной копийностью для эффективного выделения используется различный объем суспензионной культуры (Таблица 10).
Таблица 10– Выход ДНК в зависимости от копийности плазмиды
| Копийность плазмиды | Объем среды | Предполагаемый выход ДНК |
| высокая | 25–50 мл | 300–500 мкг |
| низкая | 50–150 мл | 100–350 мкг |
Для выделения плазмидной ДНК использовали реактивы из набора Plasmid Midiprep 2.0: РНКаза A (лиофилизированная), ресуспендирующий раствор, лизирующий раствор, нейтрализующий раствор, промывочный раствор (концентрат), элюирующий раствор – 5 mM Tris-HCl 7,5-pH, раствор для удаления эндотоксинов.
Также использовали следующие материалы и оборудование:
Пробирки объемом 50 мл для центрифугирования;
Микроцентрифужные пробирки на 2 мл, Eppendorf;
Штатив для пробирок на 50 мл;
Охлаждаемая центрифуга с ротором для пробирок на 50 мл, относительное центробежное ускорение не менее 3 000 g;
Настольный твердотельный термостат;
Этиловый спирт (96%).
Приготовление растворов для выделения плазмидной ДНК: во флакон с концентрированным промывочным раствором добавили 210 мл 96% этилового спирта, перемешали. Чтобы растворить «РНКазы A» добавили в пробирку 0,5 мл ресуспендирующего раствора. После растворения перенесли полученный раствор во флакон с ресуспендирующим раствором», перемешали.
Выделение ДНК осуществляли согласно протоколу «Plasmid Midiprep 2.0». Перед началом работы включили охлаждение в центрифуге; охладили нейтрализующий раствор при +4°C; для улучшения элюции нагрели флакон с элюирующим раствором до +50°C. Далее перенесли ночную бактериальную культуру в пробирку на 50 мл, осадили клетки центрифугированием (3 773 g) при температуре 4°C в течение 15 минут. Полностью удалили осветленную культуральную среду. Центрифугировали пробирку с осадком 30 секунд. Пипеткой удалили остатки культуральной среды. Добавили 4 мл ресуспендирующего раствора с «РНКазой А» к осадку и тщательно ресуспендировали на вортексе до гомогенности суспензии. Добавили 4 мл лизирующего раствора, перемешали содержимое пробирки 6‑кратным переворачиванием, пока лизат не стал вязким и полупрозрачным. Инкубировали при комнатной температуре не более 5 минут, больше 5 минут не рекомендуется, иначе плазмидная ДНК денатурирует. Добавили 4 мл предварительно охлажденного нейтрализующего раствора, перемешали содержимое, переворачивая пробирку до образования равномерной творожистой взвеси. Центрифугировали пробирку 30 минут для осветления лизата (3 773 g) при комнатной температуре. Поместили спин-колонку в пробирку на 50 мл, пробирку установили в штатив для более удобной работы. Добавили в центр спин-колонки 0,5 мл раствора для удаления эндотоксинов, чтобы уравновесить колонки. Перенесли осветленный бактериальный лизат в колонку. Центрифугировали пробирку со спин-колонкой 1 минуту не более 1850 g при комнатной температуре. В процессе центрифугирования плазмидная ДНК собирается на фильтре колонки. Далее удалите фильтрат из пробирки. В колонку добавили 2 мл раствора для удаления эндотоксинов. Центрифугировали пробирку с колонкой 1 минуту при 1 850 g при комнатной температуре. Добавили 15 мл промывочного раствора со спиртом в колонку. Центрифугировали пробирку с колонкой 1 минуту при 3 773 g при комнатной температуре. Удалили фильтрат из пробирки. Еще раз повторили процедуру: добавили 15 мл промывочного раствора со спиртом в колонку. Центрифугировали пробирку с колонкой 1 минуту при 3773g при комнатной температуре и снова удалили фильтрат из пробирки. Центрифугировали пробирку с колонкой 10 минут при 3773 g. Поместили спин-колонку в новую пробирку на 50 мл. Чтобы остатки спирта на фильтре не снизили выход ДНК и не повлияли на спектрофотометрические измерения спин-колонки посушили при комнатной температуре 8 минут. Нанесли на фильтр спин-колонки 2–3 мл предварительно нагретого до 50°C элюирующего раствора. Инкубировали 2 минуты при комнатной температуре. Центрифугировали колонку 1 минуту для сбора очищенной ДНК при 3 773 g, элюат собирается в пробирки.
Для очистки плазмидных векторов и удаления остаточных эндотоксиноввыполняли переосаждение плазмидной ДНК спиртом. Центрифугирование при переосаждении на всех этапах проводили при комнатной температуре на скорости 12 000 об/мин. Для переосаждения плазмидной ДНК использовали пробирки Eppendorf и высокоскоростную центрифугу «СМ-50» производитель компания ELMI (Латвия).
Переосаждение проводили согласно следующему протоколу. Отобрали аликвоту по 0,5 мл очищенной на спин-колонке плазмидной ДНК по двум участкам гена миостатина в микроцентрифужные пробирки объемом 2 мл (10 штук – по 5 пробирок на последовательность). Добавили 50 мкл 3M ацетата натрия pH 5,2 и 1,4 мл 96% этанола, перемешали на вортексе. Инкубировали 2 минуты при комнатной температуре. Центрифугировали образцы в настольной центрифуге 10 минут. Аккуратно удалили осветленную жидкость, чтобы не захватить на дне пробирки беловатый осадок. Аккуратно добавили 0,5 мл 70% этилового спирта. Поместили пробирку в ротор в том же положении. Центрифугировали образец в настольной центрифуге 10 минут. Аккуратно удалили спиртовую фазу и подсушили осадок при комнатной температуре 8 минут. Осадок стал полностью прозрачным (бесцветным). Растворили осадок в 50 мкл элюирующего раствора.
8. Оценка качества выделенной ДНК и полученных конструкций
Концентрацию плазмидной ДНК проверяли на приборе Nanophotometer «ImpLEN». Линзу прибора аккуратно очистили салфеткой, чтобы избежать ложноположительного или ложноотрицательного результата. В качестве бланка использовали элюирующий раствор (1,5 мкл), в котором была элюирована полученная плазмидная ДНК. На линзу нанофотометра нанесли 1,5 мкл плазмидной ДНК. Согласно инструкции, соотношение А260/280 должно быть равным 1,7-1,9 нг/мкл. Соотношение А260/230 имеет второстепенную важность, но должно вписываться в пределы 1,5-3,0 нг/мкл. Результаты фотометрии полученных образцов приведены в Таблице 11 и графически отображены на Рисунке 16.
Таблица 11– Проверка качества выделенной плазмидной ДНК
| ДНК | Конц., нг/мкл | А230 | А260 | А280 | А320 | А260/
А280 |
А260/
А230 |
| Blank (эл. р-р ) | 0,0000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 |
| Crispr Ovis 1 | 31,650 | 0,351 | 1,086 | 0,557 | 0,013 | 1,972 | 3,175 |
| Crispr Ovis 2. | 31,600 | 0,319 | 1,077 | 0,544 | 0,005 | 1,989 | 3,414 |
| Crispr Ovis 3 | 33,800 | 0,370 | 1,067 | 0,535 | 0,011 | 2,015 | 2,942 |
| Crispr Ovis 38. | 33,900 | 0,334 | 1,080 | 0,563 | 0,002 | 1,922 | 3,247 |
Согласно полученным данным, качество выделенной плазмидной ДНК, хорошее и отвечает практически всем предъявляемым критериям.
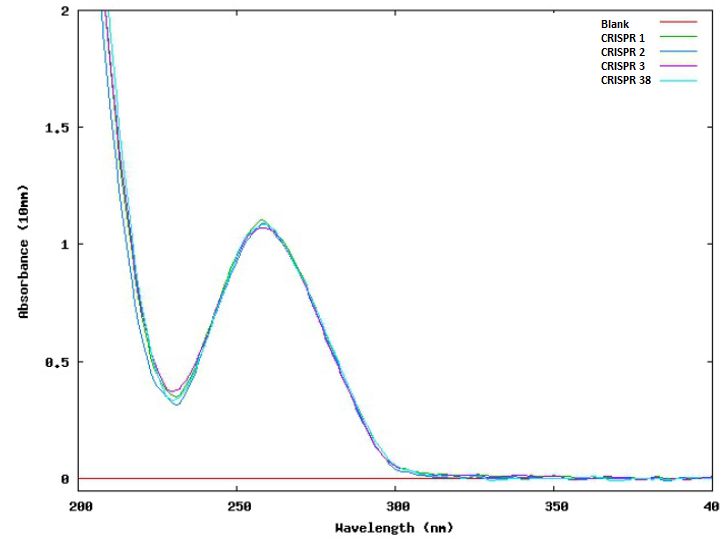
Рисунок 16. Концентрация полученной плазмидной ДНК
Однако, соотношение А260/А230 превышало желательное значениев трех пробах. В связи с этим, для дальнейшей трансфекции с плазмидными векторами, проводили очистку образцов от примесей при увеличении концентрации плазмидной ДНК.Для проверки качества переосажденной ДНК также использовали прибор Nanophotometer «ImpLEN».
Согласно полученным результатам, в результате переосажденияконцентрация плазмидной ДНК в анализируемых пробах увеличилась в 10-20 раз, что позволяет провести процедуру трансфекции (Таблица 12). По данным таблицы видно, что соотношение А260/А280 и А260/А230 вписываются в пределы нормы.
Таблица 12– Оценка качества переосажденной плазмидной ДНК
| ДНК | Конц., нг/мкл | А230 | А260 | А280 | А320 | А260/
А280 |
А260/
А230 |
| Blank (эл. р-р ) | 0,0000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 |
| Crispr Ovis 1 | 633,70 | 8,342 | 14,55 | 8,635 | 1,876 | 1,875 | 1,960 |
| Crispr Ovis 1 | 476,95 | 6,424 | 10,87 | 6,436 | 1,331 | 1,869 | 1,873 |
| Crispr Ovis 2 | 492,35 | 6,241 | 11,01 | 6,430 | 1,163 | 1,870 | 1,939 |
| Crispr Ovis 2 | 518,00 | 7,157 | 12,19 | 7,348 | 1,830 | 1,877 | 1,945 |
| Crispr Ovis 3 | 994,10 | 8,724 | 19,92 | 10,33 | 1,038 | 1,932 | 2,289 |
| Crispr Ovis 3. | 1000,6 | 11,35 | 21,49 | 11,83 | 1,179 | 1,915 | 2,194 |
| Crispr Ovis 38 | 998,40 | 11,20 | 21,52 | 11,71 | 1,385 | 1,922 | 2,206 |
| Crispr Ovis 38 | 997,70 | 11,45 | 20,24 | 11,01 | 1,389 | 1,868 | 2,386 |
Для того чтобы оценить качество полученных конструкций и убедиться, что направляющие нуклеотидные последовательность встроились в вектор в правильной ориентации и без каких-либо ошибок было проведено секвенирование плазмидных векторов выделенных из колоний клеток трансформантов.
Секвенирование нуклеазной конструкции CRISPR выполнялось с использованием прямого праймера U6, входящего в комплект GeneArt® CRISPR Nuclease Vector Kit — 5′-GGACTATCATATGCTTACCG -3′. Последовательности, полученные в ходе секвенирования векторов, выделенных из клеток трансформантов представлен в Таблице 15.
Таблица 15 – Фрагменты последовательности векторов, выделенных из различных колоний клеток-трансформантов
| Колония | Вектор |
| Crispr Ovis 1 | |
| А1 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGTTTATGCTGC TTGTTGCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| В1 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGTTTATGCTGC
— GTTGCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| Crispr Ovis 2 | |
| А2 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGTGACCGTTTCC
GTCGTAACGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| В2 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGTGACCGTTTCC
GTCGTAACGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| Crispr Ovis 3 | |
| А3 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGCGATGACTACC
ACGTTACGAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| В3 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGCGATGACTACC
—TTACGAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| Crispr Ovis 38 | |
| А38 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCGCTGTGTAATGC ATGCTTGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
| В38 | GGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTG GCTTTATATATCTTGTGGAAAGGACGAAACACCG—-GTAATGC ATGCTTGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT |
Из таблицы видно, что в колониях A1, А2, В2 А3, А38 трансформированных с использованием соответствующихвекторов последовательность анализируемыхфрагментов соответствует ожидаемой и не различается, направляющая последовательность CRISPR (в таблице выделена жирным шрифтом), имеет правильную ориентацию. В колониях В1, В3, В38анализируемый фрагмент имеет правильную ориентацию, однако содержит делеции длинной 2-4 п.н. в области направляющей последовательности и таким, образом эти колонии не могут быть использованы в дальнейшей работе. Для наращивания количества работоспособных векторов в дальнейшей работе использовались колонии A1, А2, В2 А3, А38.
9. Получение культур фибробластов, полученных от экспериментальной группы овец
Получение культуры фибробластов овцы осуществляли по общепринятым методикам [59–62].
На первом этапе проводилась биопсия кожи. С помощью лезвия выбривали участок кожи у основания ушной раковины, после чего дезинфицировали его 70 % этиловым спиртом. После дезинфекции кожу захватывали стерильными гемостатическим зажимом и скальпелем срезали кусочек кожи над браншами зажима.Отсеченные фрагменты кожи погружали во флакон с предварительно подогретым до 37 °С фосфатным буфером с добавлением антибактериального препарата пенициллин- стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина). В течение 30 минут биоптат доставлялся в лабораторию.
Дальнейшую работу вели в ламинарном боксе по следующей схеме:
-внутреннюю поверхность ламинарного бокса предварительно облучали УФ-светом и обрабатывали 70%-м раствором этилового спирта;
-полученный биоптат кожи с измельчали на фрагменты размером 1‑3 мм, отмывали от эритроцитов фосфатным буфером с добавлением антибактериального препарата пенициллин- стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина);
-для ферментативной дезагрегации ткани полученный материал инкубировали в термостате при температуре 37 °С в течение 30 мин в 0,25 % растворе трипсина;
-полученную суспензию клеток центрифугировали на скорости 2000 об. в течение 5 минут;
— супернатант переносилина дно предварительно подогретой, стерильной чашки Петри, при этом равномерно распределив его по всей поверхности. Чашку Петри, предварительно заполняли культуральной средой (4 мл). Далее чашку ставили в термостат на инкубацию при 37 °С в атмосфере, содержащей 5 % СО2.
Среду для культивирования фибробластов готовили из стерильной среды ДМЕМ с глутамином и глюкозой 4,5 г/л, с добавлением 10% эмбриональной телячьей сыворотки и антибактериального препарата пенициллин-стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина).
Каждые 48 часов (2-е суток) чашки Петри просматривали с помощью инвертированного микроскопа, с последующей оценкой качества культуры и созданием серии снимков. По мимо, контроля за ростом клеток, отслеживали контаминацию.
Культуру клеток фибробластов инкубировали в СО2–инкубаторе Binder С–150 (Binder, Германия). В ходе исследования отмечали, что на 5 сутки культивирования на дне сосуда появлялись единичные фибробласты – плоские клетки веретеновидной формы с отростками, прозрачной цитоплазмой и плоским овальным ядром.
На 8 сутки культивирования на дне чашки Петри количество прикрепленных клеток увеличилось. Еженедельно проводили замену среды в чашках Петри, для непрерывного и полноценного роста клеток(Рисунок 17).

Рисунок 17.Культура клеток на 8-е сутки
Смену среды осуществляли в ламинарном боксе по протоколу:
- Перед работой обрабатывали руки 70 % спиртовым раствором. Из чашки Петри с культурой клеток отбирали 1 мл старой среды в пробиркутипа Эппендорф и центрифугировали её 3 минуты при 2000 оборотах, 0,5 мл отбирали в новую пробирку и ставили её в твердотельный термостат, остальную среду вместе с пробиркой утилизировали;
- Оставшуюся среду из чашки Петри сливали в чашку для отходов;
- В чашку Петри наливали фосфатный буфер и промывали, сливали в чашку для отходов;
- Затем в чашку Петри добавляли 3,5 мл свежеприготовленной среды для культивирования фибробластов и 0,5 мл отцентрифугированной старой среды. Далее чашку Петри с культурой клеток переносили в термостат.
Для получения линии фибробластов, монослой пересевали, используя метод трипсинизации.
Под микроскопом просматривали чашку Петри с фибробластами. Культуру клеток с конфлюентностью свыше 60% готовили к пересеву. Работу по пересеву культуры вели в ламинарном боксе.
Пересев клеток выполняли на 14-е – 16-е сутки по следующему протоколу:
- Из чашки Петри отбирали 1 мл «старой» среды в пробирку типа Эппендорф. Центрифугировали 5 минут при 2000 оборотах, затем отбирали 0,5 мл отцентрифугированной среды и оставляли ее, для последующего переноса в культуральный матрас.
- Оставшуюся старую среду из чашки Петри сливали. В чашку Петри наливали раствор Версена, промывали культуру клеток. После промывки буфер сливали.
- В чашку Петри с культурой клеток заливали раствор трипсин 0,25 % (2,5 мл) и ставили на 5 минут в термостат (или на термостолик) при температуре 37°С. Наблюдали в окуляры микроскопа за снятием монослоя. Процесс длится 15 – 30 минут.
- Из чашки Петри раствор с клетками переносили в эппендорф пробирку.
- Центрифугировали пробирку на скорости 2000 оборотов в течение 5 минут. Удаляли надосадочную жидкость.
- Вносили в пробирку с клетками 600 мкл среды ДМЕМ. Содержимое пробирки плавно перемешали.
- Центрифугировали пробирку на скорости 2000 оборотов в течение 5 минут. Удаляли надосадочную жидкость.
- Вносили в пробирку с клетками 600 мкл среды для культивирования фибробластов. Содержимое пробирки плавно перемешали.
- Содержимое из пробирки эппендорф переносили в чашку Петри для дальнейшего культивирования.
- Затем в чашку Петри добавляли 0,5 мл старой среды (пункт 2.). Заранеев чашку Петри наливали 3 мл свежеприготовленной среды для культивирования фибробластов и ставили в термостат при температуре 37 °С. Для наращивания культуральной массы использовали не только чашки Петри, но и культуральные флаконы.
После пересева ежедневно проводили оценку роста культуры в культуральных сосудах для определения цвета среды и ее прозрачности.
Через три дня при помощи инвертированного микроскопа наблюдали большое количество адгезированных клеток на дне культуральных флаконов, которые имели вытянутую, отросчатую и веретенообразную формы с прозрачной цитоплазмой (Рисунок 18). Небольшое количество среды после каждого пересева отливали для проверки на стерильность.

Рисунок 18. Прикрепленные клетки фибробластов через 3 дня после пересева
На 5-7 сутки клетки фибробластов начинали активно делиться, тем самым увеличивая популяцию (Рисунок 19).

Рисунок 19. Прикрепленные клетки фибробластов на 9-е сутки после пересева
К 10 — 12 суткам нормальные активированные фибробласты приобретали звездчатую форму (Рисунок 20).

Рисунок 20. Фибробласты отростчатой и веретеновидной формы на 12 е сутки культивирования
Пересевы производили для получения нужного количества клеток, а также для криоконсервации.
Криоконсервацию проводили следующим образом:
- Клетки снимали с субстрата методом трипсинизации и ресуспензировали их в среде для культивирования фибробластов;
- Просчитывали количество клеток с помощью камеры Горяева. Исходную концентрацию клеток определяли по формуле 1
(1)
где N – количество клеток в 25 квадратах, V- объем суспензии;
- Осаждали клетки, центрифугируя при 2000 об. 5 минут, затем удаляли супернатант, не затрагивая клеточный осадок;
- Ресуспендировали клетки в концентрации 1*106 — 5*106 клеток/мл в 20 мкл среды ДМЕМ. Полученную суспензию переносили в среду для замораживания;
- Разливали в пробирки (2 мл пробирки с завинчивающимися крышками) для замораживания. Охлаждали пробирки в холодильнике при — 40С;
- Клетки замораживали медленно. Контейнер помещали в морозильник при -700С;
- После замерзания контейнер с пробирками переносили для хранения в жидкий азот.
Рисунок 21. Культура дермальных фибробластов овцы на 7-е сутки инкубации после оттаивания, пассаж 5
Среду для криоконсервации фибробластов готовили перед использованием, для этого смешивали 70 % эмбриональной бычьей сыворотки, 5 % DMSO, 1 % раствор антибактериального препарата пенициллин- стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина) и добавляли среду ДМЕМ с глутамином и глюкозой 4,5 г/лдо требуемого объема.
Культура клеток 5 и 6 пассажа после замораживания-оттаивания представлена на рисунке 21.Определение жизнеспособности клеток проводили с помощью окрашивания проб с культурой клеток трипановым синим. Для определения количества живых клеток 40 мкл клеточной суспензии смешивали с равным количеством 0,4% раствора красителя в 0,9 % растворе NaCl. Под покровное стекло камеры Горяева 40 микролитров смеси вносили. Количество «квадратов», в которых просчитываются клетки зависело от плотности клеточной культуры и от той точности, которая необходима (не менее 40-50 клеток). Исходную концентрацию клеток рассчитывали по формуле 1. Жизнеспособность клеток после замораживания-оттаивания в среднем составляла 94 % (рисунок 22)

Рисунок 22. Культура клеток дермальных фибробластов 6 пассажа после замораживания-оттаивания.
Полученная культура фибробластов использовалась для геномного редактирования с применением системы CRISPR/Cas9.
10. Введение разных вариантов генетических конструкций в фибробласты
Введение генетических конструкций в фибробласты выполняли с использованием двух различных методов – липофекции (биохимический) и электропорации (физический)
Химический метод трансформации клеток осуществляли с помощью липофектамина ‑Lipofectamine® 3000 Reagent.
Трансфекцию с применением липофектамина выполняли для культуры клеток, конфлюентность которой составляет 50‑90 %.
Подготовка растворов (из расчета на одну лунку):
В первой пробирке типа Эппендорф смешивали 25 мкл среды ДМЕМ с глутамином и глюкозой 4,5 г/л и 2 мкл липофектамина
Во второй пробирке смешивали 1 мкл реагента Р3000, 25 мкл среды ДМЕМ с глутамином и глюкозой и 1 мкл плазмиднойДНК CRISPR и 1 мкл ДНК донорной последовательности. ДНК предварительно размораживали и пипетировали. Содержимое этой пробирки инкубировали при комнатной температуре 15 минут. Получили ДНК-липидный комплекс.
Содержимое двух пробирок смешали, пипетировали и повторно инкубировали при комнатной температуре 15 минут.
Подготовка планшета с культурой клеток. Из лунок удаляли старую среду. В лунку с клетками добавляли 50 мкл ДНК-липидного комплекса и 150 мкл среды ДМЕМс глутамином и глюкозой 4,5 г/л. Планшет инкубировали в течение 3х часов в термостате.
Готовили 200 мкл среды ДМЕМ с глутамином и глюкозой 4,5 г/л с добавлением 10 % эмбриональной телячьей сыворотки. Готовую среду наливали в лунку с клетками и ДНК-липидным комплексом.
Оценку экспрессии генов проводили через двое суток.
Эффективность трансфекции липофектамином в среднем сооставила 28 %.
Физический метод трансфекции клеток осуществляли электропорацией. Процедуру выполняли на приборе Multiporator 4308 одиночным импульсом продолжительностью 25 мс, при напряжении 220 В и емкости 960мкФ в кюветах Gene Pulser с зазором 0,2 см.
Процедуру трансфекции выполняли по следующему протоколу:
| 1. | Работу проводили в ламинарном боксе. Перед работой обрабатывали руки 70ᴼ спиртом. |
| 2. | Из флаконов с культурой клеток отбирали 1,5 мл “старой” среды в пробирку Эппендорфа. Центрифугировали, затем отбирали 1 мл супернатанта и оставляли в твердотельном термостате. |
| 3. | Оставшуюся старую среду из флакона утилизировали. Во флакон с клетками наливали раствор Версена и промывали. |
| 4. | Во флакон с культурой клеток заливали 0,25 % раствор трипсина с ЭДТА (5мл) и ставили на 5-7 минут на термостолик инвертированного микроскопа и наблюдалив окуляры за снятием монослоя. |
| 5. | Из флакона раствор с клетками переносили в 15мл пробирку типа фалькон |
| 6. | Центрифугировали пробирку на скорости 2000 оборотов в течение 5 минут. Удаляли надосадочную жидкость. |
| 7. | Ресуспендировали осадок в 1мл PBS. Повторяли пункт 6. |
| 8. | Ресуспендировали осадок в 300 мкл буфера для электропорации GTporator . Отбирали 40 мкл раствора с клетками в пробирку эппендорф для подсчета клеток в камере Горяеева.
Подсчет осуществляли по формуле: Концентрация — С [cell/мл] = 2.5×105×(количество клеток в одном большом квадрате) Общее кол-во клеток N= 2.5×105×(количество клеток в одном большом квадрате)×V(объем суспензии, мл) |
| 9. | Далее раствор центрифугировали. Убирали надосадочную жидкость. |
| 10. | В отдельную пробирку эппендорф вносили 80-90 мкл GTporator, добавляли10 мкл плазмидной ДНКCRISPRи 5 мкл донорной донорной ДНК. ДНК предварительно размораживали и пипетировали. Содержимое пробирки ресуспендировали. |
| 11. | В пробирку с открученными клетками (пункт 9) вносили все количество раствора (пункт10), ресуспендировали. |
| 12. | Собирали все количество раствора и вливали его в кювету. Кювету вставляли в мультипоратор с выбранными режимами для электропорации- нажимали старт. |
| 13. | После этого весь раствор переносили в питательную среду на чашку Петри минимального объема (или матрас). |
| 14. | Туда же вносили 0,5 мл старой питательной среды (пункт 2). И ставили на инкубацию. |
Перед запуском электропорации суспензию клеток отстаивали в кювете 2 минуты. После электропорации суспензию оставляли в кювете 5-10 минут. Тест экспрессии генов проводили через 24 ч после электропорации.
Эффективность трансфекции фибробластов с использованием метода липофекции и реагента Lipofectamine 3000 была на 16 % выше, чем с использованием метода электропорации и составила 28 %. Использование физического метода трансфекции негативно влияло на выживаемость клеток и отдельные показатели жизнеспособности. В культурах, подвергшихся трансфекции с использованием метода электропорации отмечалось замедленее адгезии клеток и усиленная вакуолизация. Эффективность биохимической трансфекции дермальных фибробластов овцы в нашем эксперименте оказалась близка к эффективности, заявленной производителем трансфицирующего агента при работе с дермальными фибробластами человека и составила 28 %.
Таким образом, оптимальным методом для введения генетических конструкций в фибробласты в наших исследованиях стал химический метод трансфекции липофектамином.
11. Выращивание культур с модифицированным CRISPR/CAS геномом
Для получения культуры клеток с отредактированным геном, выполняли культивирование фибробластов, подвергшихся трансфекции с использованием разработанных векторов.
После учета трансфекции осуществляли пересев единичных трансфицированных фибробластов, для получения монокультуры клеток с редактированным геном.
Клетки с культуральных планшетов снимали методом трипсинизации.
1. Среду из лунки планшета отбирали пипеткой и утилизировали. Лунки с культурой клеток промывали раствором Версена и сливали.
2. В лунки планшета с культурой клеток заливали 0,25 % раствор трипсина-ЭДТА и ставили на 3- 5 минут на термостолик микроскопа при температуре 37°С и наблюдали в окуляры инвертированного микроскопа за снятием клеток.
3. Из лунок раствор с клетками переносили в пробирку Эппендорф.
4. Центрифугировали пробирку на скорости 2000 оборотов в течение 5 минут. Удаляли надосадочную жидкость.
5. Вносили в пробирку с клетками 600 мкл среды ДМЕМ. Содержимое пробирки плавно перемешали.
6. Центрифугировали пробирку на скорости 2000 оборотов в течение 5 минут. Удаляли надосадочную жидкость.
7. Вносили в пробирку с клетками 600 мкл среды для культивирования фибробластов. Содержимое пробирки плавно перемешали.
8. Содержимое из пробирки эппендорф переносили в новую чашку Петри для отбора клеток под флуоресцентным микроскопом.
9. В лунку нового планшета заливали по 450 мкл свежей среды для культивирования фибробластов с добавлением 50 мкл «старой среды».
10. Клетки с отредактированным геномом помещали в отдельные лунки планшета и ставили в термостат при стандартных условиях культивации клеток.
Полную среду (с сывороткой) готовили перед ее использованием. Среду для культивирования трансфицированных фибробластов готовили из стерильной среды ДМЕМ с глутамином и глюкозой 4,5 г/л, с добавлением 10% фетальной телячьей сыворотки и антибактериального препарата пенициллин- стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина).
Смену сред осуществляли по следующему протоколу:
1. Перед работой обрабатывали руки 70 % спиртовым раствором. Культуральную среду из лунок планшета собирали пипеткой и утилизировали;
2. Культуру в лунках планшета промывали раствором Версена;
3. Затем в лунки добавляли 450 мкл свежеприготовленной среды для культивирования фибробластов и 50 мкл заготовленной «старой» среды. Планшет с культурой клеток переносили в термостат.
Определение жизнеспособности клеток проводили с помощью окрашивания проб с культурой клеток трипановым синим. Для определения количества живых клеток 40 мкл клеточной суспензии смешивали с равным количеством 0,4% раствора красителя в 0,9 % растворе NaCl. 40 микролитров смеси вносили под покровное стекло камеры Горяева. Количество «квадратов», в которых просчитываются клетки зависело от плотности клеточной культуры и от той точности, которая необходима.
12. Оценка фенотипа модифицированных клеток
Морфометрические показатели (длина, ширина, площадь, ЯЦО) определяли на инвертированном оптическом микроскопе OLYMPUS GX – 71.
Морфометрические показателиопределяли в программе VideoTesTMaster 5.1 для Windows производства АОЗТ «ИСТА», Россия, г. Санкт-Петербург, на IBM-совместимом компьютере[63].
Для детекции эксрессии репортерного оранжевого флуоресцентного протеина, закодированного в плазмидном векторе и позволяющем судить об эффективности трансфекции культуры клеток просматривали под люменисцентным микроскопом (рисунок 45). Оранжевый флуоресцентный протеин флуоресцирует при длине волны 600 нм. С каждой культуры выполнялись снимки (в формате .jpg, размером 3136×2352 пикселей в палитре 24 бит) в случайно выбранных полях зрения при увеличении × 40, × 100, × 200, × 400 с помощью фотоаппарата OLYMPUS С 300 (Япония) (Рисунок 44).
Оценку культуры модифицированных клеток и добавление в среду антибактериальный препарат пенициллин- стрептомицин (100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина)выполняли через 48 часов, 96 часов, 216 часов после проведения трансфекции.
На вторые сутки культура была представлена клетками, прикрепившимися ко дну культурального флакона, вытянутой формы с длинными отростками и круглыми клетками – клетками в активной фазе деления.
Морфометрические показатели аутологичных дермальных фибробластов овец до и после трансфекции липофектамином представлены в Таблицах 13, 14.
| Показатели фибробластов до трансфекции (5 пассаж) | Показатели фибробластов после трансфекции липофектамином (5 пассаж) | |||
| Временной промежуток | Длина, мкм | Ширина, мкм | Длина, мкм | Ширина, мкм |
| 24 часа | 41,720±0,521* | 16,205±0,574* | 42,272±0,751* | 16,874±0,532* |
| 48 часов | 43,350±0,498* | 17,967±0,278* | 44,427±0,529* | 17,018±0,429* |
| 96 часов | 45,217±0,472* | 19,841±0,342* | 45,653±0,621* | 18,114±0,573* |
Таблица 13– Средние значения длины, ширины и площади клеток дермальных фибробластов овец в процессе культивирования
Примечание: статистическая значимость различий фибробластов: * – p <0,05
Таблица 14– Средние значения некоторых параметров дермальных фибробластов овец 5 пассаж 96 часов
| Показатели фибробластов до трансфекции (5 пассаж) | Показатели фибробластов после трансфекции липофектамином (5 пассаж) | ||
| Показатель ЯЦО (%) | Площадь, мкм | Показатель ЯЦО (%) | Площадь, мкмк |
| 0,076±0,009 | 894,9±86,7 | 0,053±0,005 | 825,3±72,2* |
В ходе работы с культурой клеток под флуоресцентным микроскопом нами сделаны описания фибробластов: клетки веретеновидной уплощенной формы с немногочисленными отростками, с помощью которых образуются межклеточные контакты друг с другом. Ядро в центре, округлое, гипохромное.
Рисунок 23. Трансфицированная культура фибробластов, вектор CRISPR 3, пассаж 5.
Рисунок 24. Трансфицированная культура фибробластов, вектор CRISPR 38, пассаж 5.
Эффективность трансфекции липофектамином в среднем сооставила 28 %. Для детекции эксрессии репортерного оранжевого флуоресцентного протеина, закодированного во вносимых плазмидном векторах и позволяющем судить об эффективности трансфекции культуры клеток также просматривали под люменисцентным микроскопом при длине волны 600 нм (рисунок 23, 24).
 На рисунке 25 представлены модифицированные клетки фибробластов веретеновидной формы с немногочисленными отростками просматриваемые под световым нвертированным микроскопом.
На рисунке 25 представлены модифицированные клетки фибробластов веретеновидной формы с немногочисленными отростками просматриваемые под световым нвертированным микроскопом.
Рисунок 25. Модифицированные клетки под инвертированным микроскопом на 5-е сутки культивирования
На рисунках26 и 27 представлены окрашенные культуры 0,4 % раствором трипанового синего до и после трансфекции. Количество живых клеток до трансфекции 97 %, после 95 %.
В ходе проведенных нами опытов каких-либо фенотипических особенностей у модифицированных клеток с отредактированным геномом не обнаружено, что говорит о том, что редактирование не приводит к появлению нецелевых мутаций, затрагивающих гены, участвующие в формировании клеточных структур.


Рисунок 26. Окрашенная культура клеток 5 пассажа после трансфекции липофектамином
Рисунок 27. Окрашенная культура клеток 5 пассажа до трансфекции липофектамином
13. Выделение ДНК из фибробластов, оценка эффективности редактирования генов
После трансфекции оценку результатов клеток проводили через 2 суток, затем, чтобы нарастить культуру перенесли ее в культуральный матрас, поставили в лабораторный инкубатор с атмосферой СО2 на 10 суток. Когда конфлюентность культуры составила 80 %, трансфицированные клетки снимали с подложки методом трипсинизации. Отмывали культуру в фосфатном буфере 2 раза. Готовые пробы были взяты на выделение ДНК.
Выделение ДНК из культуры клеток проводили с использованием набора «QIAamp DNA Mini Kit» производства компании «QIAGEN» (Германия), в специальном ламинарном боксе производитель компания LAMSYSTEMS. Набор предназначен для выделения и очистки общей ДНК (до 50кБ) из цельной и сухой крови, плазмы, сыворотки, лейкоцитов, лимфоцитов, тельных жидкостей, культур клеток, мазков, образцов ткани. Выделение основано на процедуре лизиса клеточных стенок с последующим связыванием ДНК с мембраной (силикой) в миницентрифужной колонке при соответствующей ионной силе и рН. Набор содержит протеиназу К, буферы AL, AW1, AW2 и AE – для элюции ДНК. Основные плюсы набора: быстрая очистка высококачественной ДНК; воспроизводимый и высокий выход ДНК; полное удаление примесей и ингибиторов. Для работы использовали высокоскоростную центрифугу «СМ-50» производитель компания ELMI (Латвия), низкоскоростную центрифугу- вортекс «FVL-2400N Combi-spin» производитель компания BioSan (Латвия), твердотельный термостат «CH‑100» с функцией охлаждения и нагрева производитель компания BioSan (Латвия), прибор Nanophotometer «ImpLEN» для определения концентрации ДНК, а также использовали стерильные пробирки, наконечники, пипетки и дополнительные материалы.
Выделение ДНК проводили согласно протоколу набора «QIAamp DNA Mini Kit». На дно микроцентрифужной пробирки внесли 20 мкл протеиназы К. Добавили в пробирку 200 мкл трансфицированных клеток в фосфатном буфере. Добавили к образцу 200 мкл буфера AL, тщательно перемешали содержимое пробирки с помощью вортекса в течение 15 секунд – это важно для обеспечения эффективного лизиса. Инкубировали пробирку с закрытой крышкой при 56°С в течение 10 минут. Удалили капли с крышки путем кратковременного центрифугирования на вортексе. Добавили 200 мкл этанола 96 % к образцу, снова перемешали с помощью вортекса в течение 15 секунд. Удалили капли с крышки путем кратковременного центрифугирования на вортексе. Осторожно перенесли полученную смесь в спин-колонку QIAamp Mini, избегая при этом смачивания ободка пробирки. Закрыли крышку и центрифугировали пробирку при 8000 об / мин в течение 1 мин. Если после центрифугирования лизат не опустился полностью в собирающую пробирку, необходимо провести повторное центрифугирование на более высокой скорости, до тех пор, пока колонка QIAamp Mini не опустеет. Перенесли спин-колонку QIAamp Mini в чистую собирающую пробирку объемом 2 мл, пробирку, содержащую фильтрат выбросили. Осторожно открыли спин-колонку QIAamp Mini и добавили 500 мкл буфера AW1 избегая при этом смачивания ободка пробирки. Закрыли крышку и центрифугировали пробирки при 13 000 об/мин в течение 1 минуту. Перенесли спин-колонку QIAamp Mini в чистую собирающую пробирку объемом 2 мл, пробирку, содержащую фильтрат выбросили. Осторожно открыли спин-колонку QIAamp Mini и добавили 500 мкл буфера AW2 избегая при этом смачивания ободка пробирки. Закрыли крышку и центрифугировали пробирку на полной скорости (14000 об / мин) в течение 3 минуты. Содержимое собирающей пробирки слили в емкость с отходами. Удалили остатки жидкости с ободка пробирки сухой бумажной салфеткой. Вернули колонку в очищенную собирающую пробирку. Провели дополнительное центрифугирование в течение 1 минуты на максимальной скорости для удаления остатков этанола. Перенесли спин-колонку QIAamp Mini в чистую 1,5 мл микроцентрифужную пробирку. Открыли спин-колонку QIAamp Mini и добавили 200 мкл буфера AE. Инкубировали пробирку при комнатной температуре (23° C) в течение 5 минут, а затем центрифугировали при 8000 об / мин в теч ние 1 минуты. Извлекли и выбросили колонку из микроцентрифужной пробирки. В микроцентрифужной пробирке находится очищенная ДНК, готовая для дальнейших манипуляций. На приборе «Nanophotometer» проверяли качество и концентрацию ДНК(Рисунок 23, Таблица 15).
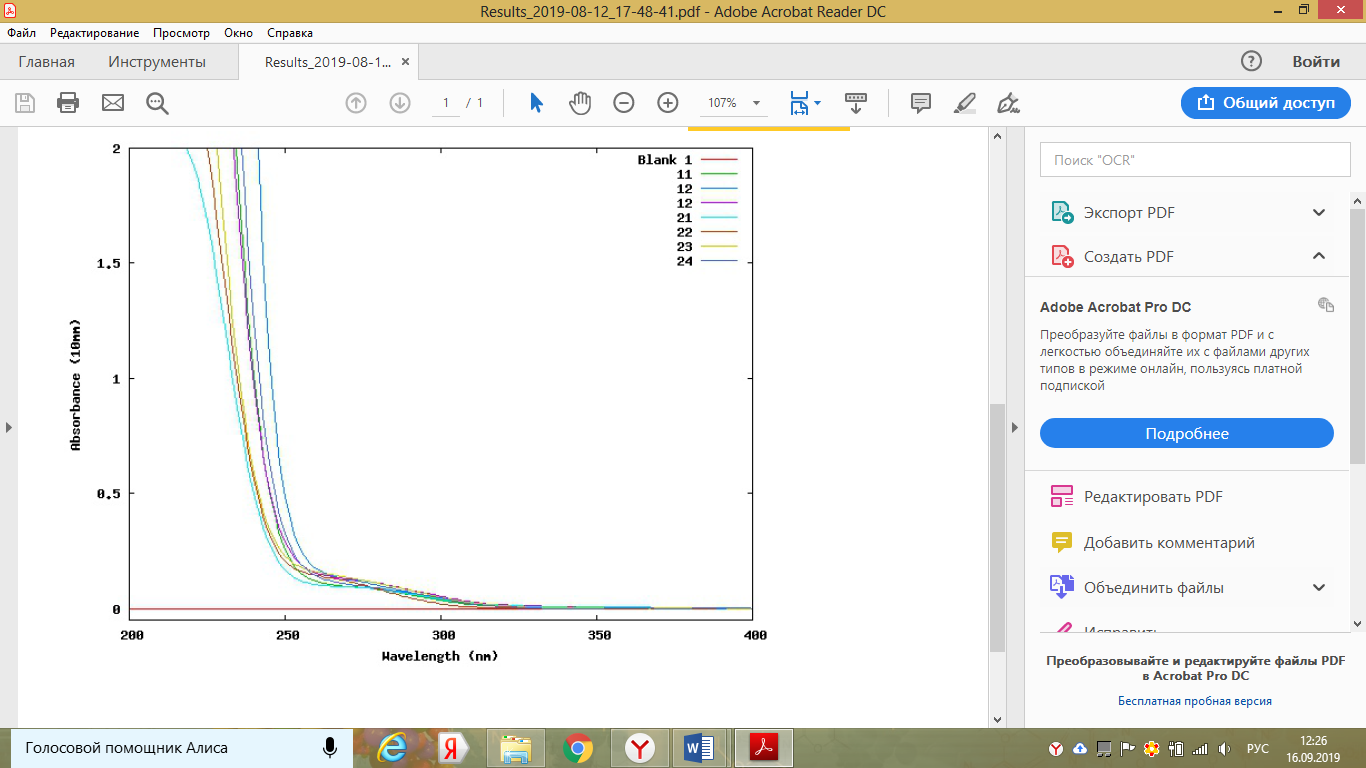
Рисунок 28. Графическое отображение результатов фотометрии ДНК, выделенной из культур клеток
Линзу прибора аккуратно очистили салфеткой, в качестве бланка использовали буфер АЕ (1,5 мкл), в котором была элюирована ДНК. На линзу нанофотометра нанесли 1,5 мкл ДНК.
Таблица 15– Проверка качества ДНК, выделенной из культур клеток
| ДНК | Конц. | Ед. | А230 | А260 | А280 | А320 | А260/
А280 |
А260/
А230 |
| Blank
(б-р АЕ ) |
0,0000 | нг/мкл | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 | 0,000 |
| 11 | 5,3500 | нг/мкл | 2,936 | 0,117 | 0,082 | 0,010 | 1,786 | 1,637 |
| 12 | 7,9000 | нг/мкл | 8,096 | 0,174 | 0,100 | 0,016 | 1,881 | 1,720 |
| 12 | 6,9500 | нг/мкл | 2,689 | 0,155 | 0,110 | 0,016 | 1,879 | 1,952 |
| 21 | 4,5000 | нг/мкл | 1,281 | 0,104 | 0,083 | 0,014 | 1,804 | 1,771 |
| 22 | 7,0000 | нг/мкл | 1,484 | 0,144 | 0,083 | 0,004 | 1,772 | 1,895 |
| 23 | 7,3000 | нг/мкл | 1,717 | 0,159 | 0,109 | 0,013 | 1,721 | 1,986 |
| 24 | 6,7000 | нг/мкл | 3,548 | 0,142 | 0,088 | 0,008 | 1,875 | 1,938 |
Согласно инструкции, соотношение А260/280 должно быть равным 1,7-1,9 нг/мкл. Соотношение А260/230 имеет второстепенную важность, но, согласно инструкции, должно вписываться в пределы 1,5-3,0 нг/мкл. Согласно полученным данным, качествовыделенной ДНК удовлетворительное и, не смотря на низкую концентрацию, пробы могут быть использованы для постановки полимеразной цепной реакции и отправлены на секвенирование. Для амплификации целевых участков использовали праймеры, представленные в Таблице 7.
Для оценки эффективности геномного редактирования использовали набор для локус-специфичной рестрикцииGeneArt® Genomic Cleavage Detection Kit, основанный на примененииэндонуклеазыT7EI. Оценку проводили по следующему протоколу.
Клетки трансфицированные векторами CRISPR/Cas9 диссоциировали 0,25 % раствором трипсина-ЭДТА, отмывали фосфатным буфером и осаждали центрифугированием при 200 g в течение 5 минут.
Осторожно удаляли супернатант и приступали к лизису клеточного осадка.Для этого готовили в стерильной микроцентрифужной пробирке лизирующую смесь, из расчета 50 мкл лизирующего буфера Cell Lysis Buffer и 2 мкл смеси для белковой деградации Protein Degrader Mix на одну пробу.
Вносили по 50 мкл лизирующей смеси в пробирки с клеточным осадком, ресуспендировали содержимое пробирки плавным пипетированием. Переносили содержимое в чистую пробирку для ПЦР.
Запускали на термоциклере программу для лизирования:
Таблица 16 – Режим лизирования
| Температура | Время | Цикл |
| 68 °С | 15 мин | 1 |
| 95 °С | 10 мин | 1 |
| 4 °С | не более 3 мин | 1 |
После завершения лизирования немедленно приступали к амплификации. Компоненты ПЦР реакции представлены в таблице 17, режим амплификации в таблице 18.
Таблица 17 – Компоненты реакционной смеси
| Компонент | Образец (мкл) | Контроль (мкл) |
| Лизирующий буфер | 2 | — |
| Смесь праймеров F/R | 1 | — |
| Контрольная матрица | — | 1 |
| AmpliTaq Gold 360 Мастермикс | 25 | 25 |
| Вода | 22 | 24 |
| Общий объем | 50 | 50 |
Таблица 18 – Режим амплификации
| Этап | Температура | Время | Цикл |
| Активация фермента | 95 °С | 10 мин | 1 |
| Денатурация | 95 °С | 30 сек | 40 |
| Отжиг | 55 °С (Tm) | 30 сек | |
| Элонгация | 72 °С | 30 сек | |
| Финальная элонгация | 72 °С | 7 мин | 1 |
| Хранение | 4 °С | Хранение | 1 |
После завершения амплификации продукт реакции анализировали с помощью электрофореза в 2 % агарозном геле для проверки размера полученных ампликонов. На гель вносили смесь, состоящую из 3 мкл продукта ПЦР и 10 мкл воды. Для сопоставления размера полученных ампликонов вносили в гель весовой маркер ДНК. На геле были выявлены полосы ожидаемого молекулряного веса –составляющие ~280 п.н.
Проводили случайный отжиг фрагментов ПЦР для формирования гетеродуплексов. В пробирку вносили 2 мкл продукта ПЦР, 1 мкл 10-кратного детектирующего буфера,6 мкл воды. Аккуратно центрифугировали. Запускали программу отжига (Таблица 19).
Таблица 19 – Режим повторного отжига
| Этап | Температура | Время | Температура/время |
| 1 | 95°С | 5 минут | – |
| 2 | 95°С–85°С | – | -2°С/сек |
| 3 | 85°С– 25°С | – | -0,1°С/сек |
| 4 | 4°С | – | Хранение |
После завершения отжига немедленно приступали к анализу расщепления.На этой стадии гетеродуплексные ДНК, содержащие инсерции или делеции подвергались расщеплению ферментом обнаружения, что позволило количественно определить долю геномных модификаций с использованием программы гелевого анализа.
В пробирки с анализируемыми образцами вносилипо 1 мкл детектирующего фермента, содержимое пробирки хорошо перемешали. Пробирки инкубировали при 37°C в течение 1 часа, затем встряхивали и осаждали на вортексе, сохраняли до проведения электрофореза при температуре 4° C. Оценивали результаты рестрикции методом гель-электрофореза. В электрофорезную кювету заливали 2% гель E-Gel EX Gel толщиной 0,4 см, осторожно удаляли гребенку. В лунки вносили по 10 мкл тестируемого образца, в одну лунку добавили контрольный образец. Кювету с гелем поместили в электрофорезную камеру на 30 минут при низком напряжении. Для анализа полученных результатов использовали гель-документирующую систему Gel Doc XR+.По результатам гель-документации ампликоны, полученные в результате амплификации ДНК клеток, отредактированных с использованием вектора Rank 1 и донорной последовательности ssODN1, после расщепления были представленытремя полосами полосами- 285 пар оснований (родительская полоса), 150 пар оснований и 135 пар оснований. Ампликоны, полученные в результате амплификации ДНК клеток, отредактированных с использованием вектора CRISPR1 и донорного вектора, после расщепления были представлены четырьмя полосами 417 пар оснований, 285 пар оснований, 160 пар оснований, 125 пар оснований.Ампликоны полученные в результате амплификации ДНК клеток, отредактированных с использованием вектора CRISPR 2 и донорной последовательности ssODN 2 после расщепления, также были представленычетырьмя полосами-285 пар оснований (родительская полоса), 150 пар оснований, 125 пар оснований, 20 пар оснований. Ампликоны полученные в результате амплификации ДНК клеток, отредактированных с использованием вектора CRISPR 3 и донорной последовательности ssODN 3 после расщепления, были представлены пятью полосами — 285 пар оснований (родительская полоса), 150 пар оснований, ~125 пар оснований, 20 и10 пар оснований. Ампликоны полученные в результате амплификации ДНК клеток, отредактированных с использованием вектора CRISPR 38 и донорной последовательности ssODN 38 после расщепления, были представлены тремяполосами — 285 пар оснований (родительская полоса), 220 пар оснований, 65 пар оснований.Данные об эффективность геномного редактирования приведены в Таблице 20. Эффективность оценивали по формуле:
% модификаций = 100 x (1 – (1- фракция расщепления)1/2)[64].
Таблица 20 – Эффективность геномного редактировании при использовании различных векторов
| Редактирующий вектор | Донорная последовательность | Эффективность, % | |
| Проба 1 | Проба 2 | ||
| CRISPR 1 | ssODN1 | 28,4 | 0,0 |
| CRISPR 2 | ssODN2 | 0,0 | 15,5 |
| CRISPR 3 | ssODN3 | 32,8 | 5,4 |
| CRISPR 38 | ssODN38 | 7,5 | 17,4 |
| CRISPR 38 | MultiLig6242 | 0,0 | 13,0 |
Полученные результаты говорят об успешно проведенном редактировании первого экзона гена миостатина.
14. Выбор наиболее эффективных вариантов генных конструкций и их доставки
По результатам секвенирования ДНК клеток, подвергшихся трансфекции, установлено, что в анализируемых пробах помимо последовательностей гена миостатина дикого типа присутствовали мутантные аллели, имеющие инсерции и делеции различной длины.
При использовании редактирующего вектора CRISPR 1в комбинации с донорной последовательностью ssODN1было получено 3 варианта мутантной последовательности, имеющие делеции и инсерции длиной 2-3 п.н. (Рисунок 24, строки Mut 1.1-Mut 1.3).При этом вариант мутантной последовательности Mut 1.3, содержал инсерцию AAT, соответствующую фрагменту донорной последовательности.

Рисунок 29. Мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 1
Использование редактирующего вектора CRISPR 1 в комбинации с донорной плазмидой MultiLig6242 позволило получить мутантную последовательность, имеющие инсерцию длиной 133 п.н. (Рисунок 24, строка Mut 1.4. Последовательность выявленной инсерции соответствовала фрагментупоследовательности зеленого флуоресцентного протеина, закодированного в донорной плазмиде. Последовательность инсерции: TGTTAACTTGTTTATTGCAGCTTATAATGGTTACAAATAAAGCAATAGCATCACAAATTTCACAAATAAAGCATTTTTTTCACTGCATTCTAGTTGTGGTTTGTCCAAACTCATCAATGTATCTTAACGCGT.
Согласно компьютерному анализу полученных мутантных последовательностей внесенные изменения должны приводить к изменению структуры кодируемого пептида (Рисунок 25).
Рисунок 30. Транслированные мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 1
Так при транслировании последовательности Mut 1.1 обнаруживается выпадение аминокислоты треонин в позиции 114, а также замена глутаминовой кислоты на лизин в позиции 116. Последовательность Mut 1.2 при аминокислотном транслировании согласно результатам анализа связана с изменением аминокислот в позициях 117-124 и укорочением кодируемого пептида на 251 аминокислоту. Последовательность Mut 1.3 при транслировании также дает укороченный пептид, при этом его длина уменьшается на 258 аминокислот, по сравнению с референсной. Последовательность Mut 1.4 при трансляции insilicoпреобразуется в пептид, состоящий из 159 аминокислот, при этом только 115 аминокислот совпадают с референсом MSTN.
При использовании редактирующего вектора CRISPR2в комбинации с донорной последовательностью ssODN2 было получено 2 варианта мутантной последовательности (Рисунок 26). Выявлена делеция длиной 6 нуклеотидов в позиции 339-344, делеция длиной 3 нуклеотида в позиции 328-330, трехнуклеотидная инсерция в позиции 337-339.
Рисунок 31. Мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 2
Согласно прогнозу трансляции внесенные изменения сопряжены с выпадением из белковой последовательности аминокислоты треонин в позициях 114-115, выпадению аспарагиновой кислоты в позиции 110 и добавлению треонина в позиции 116 (Рисунок 27). Однако, можно предположить, что подобные изменения существенно не отразятся на работе кодируемого пептида.

Рисунок 32. Транслированные мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 2
Использование редактирующего вектора CRISPR3 в комбинации с донорной позволило ssODN3 получить наибольшее количество вариантов мутантных последовательностей(Рисунок 28).Полученные последовательности содержат делеции длиной от 1 до 12 пар нуклеотидов, а также инсерции длиной 3 и 6 пар нуклеотидов. Вариант Mut 3.1 демонстрирует успешную интеграцию добавочного стоп-кодона, соответствующего донорной последовательности.
Рисунок 33. Мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 2
Внесенные изменения, согласно компьютерному анализу, серьезно отражаются на аминокислотной последовательности кодируемого пептида. Варианты Mut 3.1, Mut 3,2 и Mut 3.4 связаны с укорачиванием кодируемого белка на 281, 268 и 287 аминокислот соответственно. Вариант Mut 3.3 и Mut 3.5, согласно прогнозу, при транслировании также приводят к формированию укороченного белкового продукта, но при этом отличие от референсной последовательности длиной всего в 2-6 аминокислот.
Рисунок 34. Транслированные мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 3
При использовании редактирующего вектора CRISPR38в комбинации с донорной последовательностью ssODN38 было получено 3 варианта мутантной последовательности, имеющие делецию длиной 10 пар нуклеотидов и инсерции длиной 1-2 п.н. (Рисунок 30).
Рисунок 35. Мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 38
Внесенные изменения, согласно компьютерному анализу, привели к формированию преждевременных стоп-кодонов в позициях 42, 49 и 50 (Рисунок 31). Таким образом длина мутантного пептида по сравнению с каноническим сокращается в 7,9-9,4 раза, что при условии гомозиготной мутации должно приводить к полному нокауту гена MSTN.

Рисунок 36. Транслированные мутантные последовательности гена MSTN, полученные при использовании редактирующего вектора CRISPR 3
Согласно полученным данным, и с учетом эффективности геномного редактировании при использовании различных векторов, наиболее эффективным вариантом генных конструкций для нокаута гена миостатина при негомологичного соединении концов являются векторы CRISPR3 и CRISPR38. Для запускамеханизма гомологичной рекомбинации с целью интеграции добавочных стоп кодонов наиболее предпочтительным является использование редактирующего вектора CRISPR3в сочетании с донорной последовательностьюssODN3. Удовлетворительные результаты также получены с использованием редактирующего вектора CRISPR1в сочетании с донорной последовательностью ssODN1. Использование с целью интеграции в область гена миостатина последовательности зеленого флуоресцентного белка донорной плазмида MultiLig6242в сочетании с редактирующим вектором CRISPR 1 оказалось неэффективным, поскольку удалось добиться интеграции только небольшого отрезка хвостовой последовательности гена GFP (133 п.н.). Наиболее эффективным способом доставки используемых редактирующих конструкций в фибробласты овец, согласно проведенным исследованиям, является метод биохимической трансфекции – липофекции. Эффективность трансфекции фибробластов с использованием метода липофекции и реагента Lipofectamine 3000 была на 16 % выше, чем с использованием метода электропорации и составила 28 %.
Список использованной литературы